Lekársky expert článku

Nové publikácie
Alkaptonúria - vrodená enzymatická patológia
Posledná kontrola: 23.04.2024

Všetok obsah iLive je lekársky kontrolovaný alebo kontrolovaný, aby sa zabezpečila čo najväčšia presnosť faktov.
Máme prísne smernice týkajúce sa získavania zdrojov a len odkaz na seriózne mediálne stránky, akademické výskumné inštitúcie a vždy, keď je to možné, na lekársky partnerské štúdie. Všimnite si, že čísla v zátvorkách ([1], [2] atď.) Sú odkazmi na kliknutia na tieto štúdie.
Ak máte pocit, že niektorý z našich obsahov je nepresný, neaktuálny alebo inak sporný, vyberte ho a stlačte kláves Ctrl + Enter.
Jedna z veľmi zriedkavých metabolických porúch - alkaptonúria - sa týka vrodených abnormalít v metabolizme aminokyseliny tyrozínu.
Tento syndróm sa tiež môže nazývať nedostatok homogentisát oxidázy, homogentisinúria, dedičná ochronóza alebo ochorenie čierneho moču. [1]
Epidemiológia
Podľa štatistík nie je viac ako deväť prípadov alkaptonúrie na 1 milión populácie. A vo väčšine európskych krajín - jeden prípad na 100 - 250 tisíc živonarodených detí.
Spomedzi európskych krajín je výnimkou Slovensko (obzvlášť relatívne malý severozápadný región), kde je prevalencia alkaptonúrie jedným prípadom u 19 tisíc novorodencov. S najväčšou pravdepodobnosťou je to spôsobené tým, že v rodinách slovenských Rómov, ktorí tam žijú, je úroveň príbuzenského kríženia (manželstvá medzi bratrancami a sestrami) najvyššia v Európe: 10-14%. [2]
Príčiny alkaptonúria
Presné príčiny alkaptonúrie ako vrodenej poruchy katabolizmu (metabolického rozkladu) aromatického (homocyklického) a-aminokyselinového tyrozínu boli stanovené: tento typ metabolickej poruchy je dôsledkom homozygotných alebo komplexných heterozygotných mutácií jednej z tisícov génov na chromozóme 3, presnejšie, génu HqG21 v mieste 3 -q23 na dlhom ramene chromozómu. Tento gén kóduje nukleotidové sekvencie pečeňového enzýmu homogentisát-1,2-dioxygenázy [3](tiež nazývanej oxidáza homogentisovej kyseliny alebo homogentisát oxidázy), metaloproteínu obsahujúceho železo, ktorý je potrebný pre jeden zo štádií rozpadu tyrozínu v tele. [4], [5]
Alkaptonúria je teda defektom enzýmu homogentisát-1,2-dioxygenázy, presnejšie dôsledkom jeho geneticky podmieneného nedostatku alebo úplnej absencie. [6]
Ako vrodená fermentopatia je alkaptonúria dedičná ako autozomálne recesívny znak, to znamená, že aby sa alkaptonúria vyskytla u detí, obaja rodičia musia mať upravený gén pre tento enzým, pretože každý z nich prenáša na dieťa iba jednu kópiu gén z dvoch dostupných.
Podľa najnovších údajov existuje viac ako dvesto variantov modifikácie génu HGD a najčastejšie sa pozorujú mutácie missense, translokácia a zostrih.
Rizikové faktory
Jediným rizikovým faktorom vzniku tejto vrodenej fermentopatie je jej rodinná anamnéza a dedičnosť dvoch modifikovaných kópií génu HGD, ak rodičia nevykazujú alkaptonúriu (riziko prenosu anomálie je 25%) alebo jednu z rodičia majú túto poruchu. [7]
Patogenézy
Najdôležitejšiu úlohu zohráva tyrozín pri syntéze bielkovín, produkcii chromoproteínov - kožného pigmentu melanínu, ako aj hormónov štítnej žľazy a katecholamínových neurotransmiterov.
Mechanizmus regulácie množstva tyrozínu v bunkách je veľmi zložitý a telo normalizuje svoj nadbytočný obsah štiepením. Proces katabolizmu tyrozínu, ako všetkých aromatických aminokyselín, je viacstupňový a prebieha v niekoľkých fázach. Navyše, každý stupeň metabolického rozkladu tyrozínu prebieha za účasti určitého enzýmu a tvorby medziproduktu.
Aminokyselina sa teda najskôr štiepi na para-hydroxyfenylpyruvát, ktorý sa zmení na alkapón-2,5-dihydroxyfenyloctovú alebo homogentizovú kyselinu. Ďalej by mal byť alkapón transformovaný na kyselinu maleátovú, ale to sa nestáva. [8]
A patogenéza alkaptonúrie spočíva v zastavení biochemických reakcií tyrozínového katabolizmu v štádiu tvorby kyseliny homogentisovej: na jej odbúranie jednoducho neexistuje potrebný enzým - homogentisát oxidáza.
Telo kyselinu homogentisovú nevyužíva a môže sa hromadiť pri vylučovaní obličkami. Okrem toho sa oxiduje na benzochinoacetát (kyselina benzochinonoctová), ktorý väzbou na molekuly tkanív a telesných tekutín vytvára biopolymérne zlúčeniny sfarbené ako melanín.
Akumulácia týchto medziproduktov v tkanive vedie k narušeniu štruktúry kolagénu chrupavkového tkaniva, v dôsledku čoho klesá jeho elasticita - s výskytom mnohých klinických príznakov alkaptonúrie a vznikom komplikácií.
Príznaky alkaptonúria
Alcaptonúria u novorodencov a dojčiat sa prejavuje stmavnutím moču. Pri kontakte so vzduchom sa farba moču na plienkach, plienkach a spodnej bielizni zmení na tmavohnedú; je to spôsobené akumuláciou a uvoľňovaním kyseliny homogentizovej, ktorá sa oxiduje na benzochinoacetát. [9]
Pri absencii iných symptómov nie je alkaptonúria u detí v ranom veku včas rozpoznaná, pretože po močení môže moč po niekoľkých hodinách stmavnúť. Podľa niektorých správ je v podmienkach kliník zistená iba jedna pätina detí mladších ako 12 mesiacov, ktoré sa narodili s touto fermentopatiou. Preto je veľmi dôležité, aby rodičia venovali starostlivosti o dieťa.
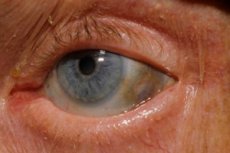
Počiatočné príznaky navyše zahrnujú pigmentáciu (modrasto sivú) očnej skléry a chrupavku ušných boltcov a nosa, často označovanú ako ochronóza. [10]
Časom sa objavia ďalšie príznaky:
- silná pigmentácia kože na lícnych kostiach, podpazuší a genitáliách;
- farbenie odevov pri kontakte s potiacimi sa časťami tela;
- záchvaty všeobecnej slabosti;
- zachrípnutý hlas.
Je potrebné mať na pamäti, že alkaptonúria a ochronóza , ako je uvedené vyššie, sú synonymné názvy pre rovnaké porušenie tyrozínového katabolizmu.
Ochorenie javorového sirupu a alkaptonúria. Vrodená choroba javorového sirupu alebo leucinóza sa tiež týka metabolických porúch, má rovnaký typ dedičnosti a dokonca sa vyskytujú mutácie na tom istom chromozóme, ale týkajú sa génu kódujúceho komplex enzýmov rozvetvenej a-keto kyseliny dehydrogenázy. Z tohto dôvodu telo nemôže rozkladať niektoré proteínové zložky, najmä aminokyseliny leucín, izoleucín a valín. V tomto stave má moč (a ušný maz) sladkú vôňu; okrem toho je v klinickom obraze tohto typu organickej acidémie pozorovaná hypopigmentácia, kolísanie krvného tlaku, kŕče, vracanie a hnačka, pokles hladiny glukózy v krvi, ketoacidóza, halucinácie atď. Úmrtnosť u detí je pomerne vysoká vysoká; u dospelých bez liečby môže v dôsledku mozgového edému nastať kóma a smrť.
Albinizmus a alkaptonúria „spájajú“ iba tyrozín. Albinizmus , vrátane oculocutaneous albinizmus, je spôsobená tým, genetické mutácie, ktoré ovplyvňujú produkciu pigment melanín. Vrodené zmeny sú zaznamenané v géne TYR na chromozóme 11 (11q14.3), ktorý kóduje tyrozinázu, enzým obsahujúci meď v melanozómoch, ktorý je potrebný na tvorbu kožného pigmentu na základe produktov metabolizmu tyrozínu. Táto choroba je oveľa bežnejšia ako alkaptonúria.
Komplikácie a následky
Následky a komplikácie alkaptonúrie spôsobené pôsobením medziproduktových metabolitov tyrozínu - homogentizových a benzochinonoctových kyselín - sa objavujú v dôsledku ukladania reaktívnych pigmentovaných polymérov, deštrukcie kolagénových fibríl a zhoršenia stavu chrupavky (so znížením ich odolnosti) na mechanické namáhanie).
O niekoľko rokov neskôr sa v dospelosti vyvíja degeneratívna artritída a artróza veľkých kĺbov (bedra, sakroiliak a koleno); zúženie medzistavcových priestorov (najmä bedrovej a hrudnej chrbtice) - s kalcifikáciou a tvorbou osteofytov; hustota tkaniva subchondrálnych kostných platničiek klesá a kosti pod nimi môžu prechádzať patologickou prestavbou s tvorbou výrastkov a deformácií. [11]
V dôsledku rovnakej kalcifikácie môže dôjsť k poškodeniu srdcových chlopní (aortálnych a mitrálnych) a koronárnych artérií so známkami koronárnej choroby srdca, ako aj k tvorbe kameňov v obličkách a prostate. [12], [13]
Diagnostika alkaptonúria
Diagnóza vrodených metabolických porúch je zvyčajne založená na štúdiu telesných tekutín.
Na základe akých testov a akých reakcií je možné diagnostikovať alkaptonúriu? Vyžadujú sa testy moču - na detekciu kyseliny homogentizovej a stanovenie jej hladiny (normálne - 20-30 mg denne, zvýšené - 3-8 g). Vzorka moču sa vyšetruje plynovou chromatografiou alebo hmotnostnou spektrometriou pomocou kvapalinovej chromatografie; je možný skríningový test na prítomnosť chloridu železitého v moči. [14]
Existuje aj rýchla diagnostická metóda - stanovenie alkaptonu v zaschnutých škvrnách moču na papieri (podľa intenzity farby).
Pri objasňovaní diagnózy zahŕňa inštrumentálna diagnostika (röntgen) identifikáciu röntgenových znakov osteoartrózy a iných kĺbových patológií u pacientov.
Diagnóza je potvrdená takými& molekulárno -genetickými metódami, ako je diagnostika dedičných chorôb , ako je genetické testovanie a sekvenovanie DNA. [15]
Odlišná diagnóza
Diferenciálna diagnostika zahŕňa hemochromatózu a akútne zlyhanie pečene novorodencov, melaninúriu, akútnu intermitentnú porfýriu, hemofagocytovú lymfohistiocytózu, primárnu mitochondriálnu patológiu, reumatoidnú artritídu, ankylozujúcu spondylitídu.
Komu sa chcete obrátiť?
Liečba alkaptonúria
Hlavnou liečbou alkaptonúrie je požitie veľkých dávok (najmenej 1 000 mg denne) kyseliny askorbovej. U detí to zvyšuje vylučovanie kyseliny homogentizovej močom a u dospelých znižuje hladinu jej derivátu, kyseliny benzochinonoctovej, v moči a spomaľuje jej väzbu na štruktúry spojivového tkaniva kĺbov a kolagénu. [16]
Na západoeurópskych klinikách sa testuje liek Nitizinone (Orfalin), liek zo skupiny metabolitov, ktorý inhibuje druhý stupeň katabolizmu tyrozínu: transformáciu para-hydroxyfenylpyruvátu na kyselinu homogentizovú. Použitie tohto farmakologického činidla však vedie k akumulácii tyrozínu a môže spôsobiť vážne vedľajšie účinky vrátane zakalenia rohovky a fotofóbie, krvácania z nosa a žalúdka, zlyhania pečene, zmien v krvi atď. V USA však Nithizinone je schválený FDA na liečbu tyrozinémie typu I. [17], [18]
Fyzioterapia - cvičebná terapia na zvýšenie svalovej sily a zvýšenie pohyblivosti kĺbov, balneo a peloidoterapia na obmedzenie bolesti - sa vykonáva pri problémoch s kĺbmi spôsobených alkaptonúriou.
Aj keď sa tyrozín nedodáva iba s jedlom, ale sa tiež vyrába v tele, pacientom s alkaptonúriou sa odporúča diéta s nízkym obsahom bielkovín a obmedzujúca príjem potravín bohatých na tyrozín, predovšetkým hovädzie a bravčové mäso, mliečne výrobky (najmä syry), strukoviny, orechy atď.
Prevencia
Prevencia génových mutácií je nemožná a aby sa zabránilo narodeniu detí s vysokým rizikom vrodených porúch, existuje lekárske a genetické poradenstvo, ktoré je potrebné pred plánovaným tehotenstvom tých manželských párov, v ktorých rodinnej anamnéze sa vyskytujú dedičné choroby. Príroda. [19]
Predpoveď
V priebehu alkaptonúrie je len veľmi málo úmrtí a smrť môže byť spôsobená vážnymi komplikáciami postihujúcimi srdce a obličky. Takže pre celkovú dĺžku života ľudí s alkaptonúriou je prognóza priaznivá.
Ale kvalita života je znížená kvôli intenzívnej bolesti kĺbov alebo chrbtice s výrazným obmedzením pohyblivosti, často progresívnej.