Lekársky expert článku

Nové publikácie
Fenylpyruvínová oligofrénia alebo fenylketonúria
Posledná kontrola: 05.07.2025

Všetok obsah iLive je lekársky kontrolovaný alebo kontrolovaný, aby sa zabezpečila čo najväčšia presnosť faktov.
Máme prísne smernice týkajúce sa získavania zdrojov a len odkaz na seriózne mediálne stránky, akademické výskumné inštitúcie a vždy, keď je to možné, na lekársky partnerské štúdie. Všimnite si, že čísla v zátvorkách ([1], [2] atď.) Sú odkazmi na kliknutia na tieto štúdie.
Ak máte pocit, že niektorý z našich obsahov je nepresný, neaktuálny alebo inak sporný, vyberte ho a stlačte kláves Ctrl + Enter.
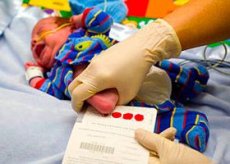
Podľa štatistík je u jedného z 10 – 15 tisíc novorodencov diagnostikovaná geneticky podmienená neurokognitívna patológia – fenylpyruvová oligofrénia, ktorá sa vyvíja v dôsledku vrodenej poruchy metabolizmu esenciálnej aminokyseliny fenylalanínu.
Toto ochorenie prvýkrát identifikoval v 30. rokoch 20. storočia v Nórsku lekár Ivar Foelling, ktorý ho nazval hyperfenylalaninémiou. V súčasnosti sa táto patológia bežne nazýva fenylketonúria, jej kód v ICD 10 je E70.0.
 [
[ Príčiny fenylpyruvovej oligofrénie
Genetické príčiny fenylpyruvickej oligofrénie sú recesívne dedičné u dieťaťa od rodičov, ktorí sú nositeľmi dvoch mutovaných alel enzýmu fenylalanínhydroxylázy (fenylalanín-4-monooxygenáza). V dôsledku toho dieťaťu úplne alebo čiastočne chýba tento enzým, ktorý je nevyhnutný na rozklad fenylalanínu (proteinogénnej kyseliny α-amino-β-fenylpropiónovej) na tyrozín [kyselina 2-amino-3-(4-hydroxyfenyl)propánová].
Genetici zistili, že patogenéza tohto ochorenia je spojená s viac ako 500 rôznymi homozygotnými alebo heterozygotnými štrukturálnymi mutáciami v chromozóme 12 - v génoch pečeňového enzýmu fenylalanínhydroxylázy 12q23.2 alebo 12q22-q24.1. Génová mutácia vedie k tomu, že hladina tohto enzýmu, nevyhnutného na oxidáciu fenylalanínu obsiahnutého v bielkovinách potravín, sa znižuje 4 alebo viackrát.
V dôsledku toho dochádza na jednej strane k hromadeniu nadbytočného fenylalanínu v krvi, ktorý je toxický pre vývoj mozgu. V tomto prípade sa fenylalanín, ktorý sa nedá premeniť na tyrozín, rozkladá deamináciou za vzniku kyseliny fenylpyrohroznovej (kyselina 2-oxo-3-fenylpropiónová alebo fenylpyruvát). Táto kyselina sa následne premieňa na kyselinu fenyloctovú (fenylacetát) a kyselinu fenylkátovú (fenyllaktát), ktoré majú negatívny vplyv na bunky mozgu a centrálneho nervového systému.
Na druhej strane, primárne príčiny fenylpyruvickej oligofrénie (vrodený nedostatok fenylalanínhydroxylázy a jeho dôsledok - nadbytok fenylalanínu) vedú k zníženiu hladín iných aminokyselín v mozgu (tryptofán, treonín, metionín, valín, izoleucín, leucín), čo úplne narúša proces biosyntézy neurotransmiterov prenášajúcich nervové impulzy - dopamínu a norepinefrínu - a spôsobuje progresívne zhoršenie duševného a fyzického vývoja u detí.
Príznaky fenylpyruvickej oligofrénie
Nadbytok fenylalanínu v tele sa prejavuje u dojčiat a jeho prvé príznaky sú: letargia a ospalosť; problémy so saním; kŕče; vracanie; ekzémové lézie epidermy; zatuchnutý (myší) zápach tela, moču a dychu (v dôsledku zvýšeného obsahu fenylacetátu, ktorý sa oxiduje na fenylketóny).
Pozorujú sa nasledujúce typické príznaky fenylpyruvovej oligofrénie: spomalenie rastu; svalová hyperkinéza, tremor alebo znížený svalový tonus; psychotické záchvaty sprevádzané nemotivovanou podráždenosťou a hnevom; znížené intelektuálne schopnosti alebo mentálna retardácia.
Deti s fenylketonúriou majú tiež hypopigmentáciu - výrazne svetlejšiu a tenšiu farbu pokožky, vlasov a očí v porovnaní s ich rodičmi. Bezprostrednou príčinou tohto príznaku je nedostatok tyrozínu, ktorý po oxidácii v melanocytoch epidermy tvorí pigment melanín.
Ako poznamenali odborníci v oblasti detskej psychiatrie, klinický obraz fenylpyruvovej oligofrénie závisí od stupňa poškodenia mozgových buniek a dieťa s touto patológiou sa môže počas prvých mesiacov života zdať normálne. Ak sa ochorenie nezistilo ihneď po narodení a dieťa nedostávalo dlhodobú liečbu, následky sú nasledovné: nezvratné poškodenie mozgu a jeho komplikácie vo forme ťažkej alebo hlbokej mentálnej retardácie (imbecilita alebo idiocia).
A potom sa môžu vyskytnúť nasledujúce príznaky fenylpyruvickej oligofrénie: nízky vzrast, kraniofaciálne abnormality (mikrocefália, vyčnievajúca horná čeľusť, široko rozmiestnené zuby, zhoršený vývoj zubnej skloviny), zvýšený svalový tonus a šľachové reflexy, hyperaktivita, zlá koordinácia pohybov a nešikovná chôdza, zvýšená podráždenosť a ďalšie behaviorálne a neurologické problémy až po duševné poruchy.
Diagnóza fenylpyruvovej oligofrénie
Včasná diagnostika fenylpyruvovej oligofrénie a rýchla liečba sú mimoriadne dôležité, pretože znižujú riziko vzniku mentálnej retardácie z 80 – 90 % na 6 – 8 % (podľa CLIMB – Národného informačného centra pre metabolické ochorenia, Spojené kráľovstvo).
Na tento účel by sa mal u dojčiat vykonať neonatálny skríning na fenylketonúriu tretí až štvrtý deň po narodení (vo výnimočných prípadoch medzi 5. a 8. dňom) pomocou mikrobiologického krvného testu na obsah fenylalanínu – tzv. Guthrieho testu.
Biochemické testy moču na fenylpyruvát tiež odhaľujú fenylketonúriu u novorodencov, ale tento test by sa mal robiť až 10 – 12 dní po narodení. Preto je medzinárodným diagnostickým štandardom stanovenie koncentrácie fenylalanínu v krvnej plazme.
Moderná inštrumentálna diagnostika založená na nových technológiách, ako je tandemová hmotnostná spektrometria, nám umožňuje odhaliť mnohé vrodené metabolické poruchy, ktoré vedú k mentálnemu postihnutiu. To zabezpečuje diferenciálnu diagnostiku genetických metabolických patológií vrátane: galaktozémie, ketonúrie (poruchy metabolizmu leucínu, izoleucínu a valínu), homocystinúrie, glutárovej acidúrie, izovalérovej acidémie, kosáčikovitej anémie, tyrozinémie, vrodenej hypotyreózy atď.
Komu sa chcete obrátiť?
Liečba fenylpyruvovej oligofrénie
Diétna terapia je prakticky jedinou účinnou liečbou fenylpyruvovej oligofrénie, ktorá umožňuje normálny vývoj mozgu. Pri začatí liečby u detí starších ako tri alebo štyri roky však diétna terapia neodstráni príznaky už vzniknutej patológie.
Dojčatá s deficitom fenylalanínhydroxylázy nemôžu konzumovať materské mlieko a existujú pre ne špeciálne mliečne zmesi bez fenylalanínu – Lofenalac, Afenilak, Aminogran, Milupa pku1 (pku2 a pku3), Periflex Infanta, XP Maxamum.
Diéta vylučuje konzumáciu mäsa, rýb, hydiny, mlieka, vajec, syra, tvarohu, zmrzliny, strukovín, orechov a mnohých ďalších produktov obsahujúcich bielkoviny. Ako náhrada zakázaných produktov na udržanie rovnováhy bielkovín v tele sa používa kazeínový hydrolyzát - zmes aminokyselín bez fenylalanínu extrahovaná z mliečnej bielkoviny (lieky Berlofan alebo Ipofenát).
Ak sa skorší lekári domnievali, že užívanie zmesí bez fenylalanínu a dodržiavanie špeciálnej diéty by malo pokračovať až 10 rokov (teda až do ukončenia procesov myelinácie mozgu), teraz je známe, že ukončenie liečby môže viesť k intenzívnejšiemu priebehu fenylpyruvovej oligofrénie a nezvratným následkom. Zatiaľ čo pacienti, ktorí dodržiavajú diétne obmedzenia, nevykazujú žiadne príznaky.
V súčasnosti sú lieky na liečbu fenylpyruvovej oligofrénie (fenylketonúrie) zastúpené liekom Kuvan na báze sapropterín dihydrochloridu - syntetickej náhrady kofaktora enzýmu fenylalanínhydroxylázy tetrahydrobiopterínu (BH4). Tablety Kuvan rozpustené vo vode sa užívajú jedenkrát denne - ráno, počas jedla. Dávka sa vypočíta individuálne - podľa telesnej hmotnosti pacienta (10 mg na kilogram). Medzi vedľajšie účinky lieku patrí vodnatý výtok z nosa, upchatý nos, bolesť hlavy, bolesť hrtana, vracanie, hnačka a občas - bolesť brucha. Užívanie tohto lieku neruší antifenylalanínovú diétu. V návode na použitie lieku sa uvádza, že jeho použitie u detí mladších ako štyri roky nebolo skúmané.
V tomto prípade sa liečba fenylpyruvickej oligofrénie vykonáva s neustálym monitorovaním hladiny fenylalanínu v krvi.
Fenylpyruvová oligofrénia alebo fenylketonúria je dedičné ochorenie a nedá sa jej predísť. Pri plánovaní tehotenstva je však možná prevencia - enzýmový krvný test a genetický test na zistenie, či sú budúci rodičia nositeľmi mutantného génu. Testy fenylalanínu v krvi sa môžu vykonať aj počas tehotenstva.
Ako píše časopis Journal of Inherited Metabolic Disease, „prognóza pre pacientov s touto patológiou, ak sa nelieči, je maximálna dĺžka života až 30 rokov v stave osoby so zdravotným postihnutím s ťažkým poškodením mozgových funkcií (pretože hladina fenylalanínu v krvi sa časom zvyšuje) a s liečbou – do staroby, s vyšším vzdelaním a úspešnou kariérou.“