Lekársky expert článku

Nové publikácie
Angelmanov syndróm u detí a dospelých
Posledná kontrola: 04.07.2025

Všetok obsah iLive je lekársky kontrolovaný alebo kontrolovaný, aby sa zabezpečila čo najväčšia presnosť faktov.
Máme prísne smernice týkajúce sa získavania zdrojov a len odkaz na seriózne mediálne stránky, akademické výskumné inštitúcie a vždy, keď je to možné, na lekársky partnerské štúdie. Všimnite si, že čísla v zátvorkách ([1], [2] atď.) Sú odkazmi na kliknutia na tieto štúdie.
Ak máte pocit, že niektorý z našich obsahov je nepresný, neaktuálny alebo inak sporný, vyberte ho a stlačte kláves Ctrl + Enter.

Existuje množstvo chorôb, pri ktorých výrazy ako „staraj sa o seba a neochorieš“ znejú prinajmenšom smiešne. Ide o patológie, pri ktorých sú niektoré duševné a fyzické abnormality vlastné detskému telu ešte pred narodením, ale rodičia za to nemôžu. Takéto choroby sú spôsobené mutáciami alebo abnormalitami v chromozómových súboroch a nazývajú sa chromozomálne alebo genetické. Angelmanov syndróm, Downov syndróm, Patauov syndróm, Edwardsov syndróm, Turnerov syndróm, Prader-Williho syndróm - to je len časť genetických chorôb z pomerne slušného zoznamu.
Syndróm šťastného muža
Tentoraz si povieme o patológii pomenovanej po anglickom pediatrovi Harrym Angelmanovi, ktorý prvýkrát nastolil túto problematiku v roku 1965, keď sa deň predtým vo svojej praxi stretol s tromi nezvyčajnými deťmi, ktoré spájali spoločné zvláštne príznaky. Lekár tieto deti nazval bábikovými deťmi a napísal o nich článok, ktorý sa pôvodne volal „Deti-bábky“. Samotný článok a jeho názov boli napísané pod dojmom z obrazu, ktorý bol videný v jednom z múzeí vo Verone. Obraz zobrazoval smejúceho sa chlapca a volal sa „Bábkový chlapec“. Spojenie dieťaťa zobrazeného na obraze s tromi deťmi, s ktorými sa Angelman kedysi stretol vo svojej praxi, podnietilo pediatra k tomu, aby deti zlúčil do jednej skupiny kvôli chorobe, ktorú mali.
Nie je ničím prekvapujúcim, že deti spomenuté v článku si iní lekári nevšimli. Veď na prvý pohľad sa zdalo, že majú úplne odlišné ochorenia, taký odlišný bol celkový klinický obraz ochorenia v 3 rôznych prípadoch. Možno by „nová“ chromozomálna patológia zaujala aj iných vedcov, ale v tom čase genetika ešte nebola dostatočne rozvinutá na to, aby potvrdila hypotézu anglického lekára. Preto bol článok po určitom záujme oň na dlhý čas odložený.
Ďalšia zmienka o Angelmanovom syndróme, ako sa teraz nazýval článok anglického pediatra G. Angelmana, pochádza zo začiatku 80. rokov 20. storočia. Až v roku 1987 sa podarilo nájsť dôvod, prečo sa malá časť detí rodí s takými odchýlkami, že zvonku sa zdajú byť neustále usmievavé a šťastné. V skutočnosti to vôbec nie je pravda a úsmev je len grimasa, za ktorou sa skrýva nešťastná ľudská duša a bolesť rodičov.
Epidemiológia
Podľa štatistík sa chromozomálna mutácia u dieťaťa môže vyvinúť na pozadí podobných mutácií u rodičov aj v ich neprítomnosti. Neexistuje jasná dedičná povaha Angelmanovho syndrómu (AS), ale pravdepodobnosť vzniku patológie u rodičov s chromozomálnymi mutáciami je pomerne vysoká.
Zaujímavé je aj to, že ak už rodina má dieťa s AS, existuje jednopercentná šanca, že sa jej narodí druhé dieťa s rovnakou poruchou, a to aj v prípade, že rodičia sú zdraví.
Stále neexistujú presné štatistiky o počte pacientov s Angelmanovým syndrómom. Možno dôvodom je rozmanitosť symptómov, ktoré sa môžu vyskytovať v určitom zložení alebo sa dlhodobo vôbec nevyskytovať. Predpokladá sa, že prevalencia ochorenia je: 1 dieťa na 20 000 novorodencov. Toto číslo je však veľmi približné.
Príčiny Angelmanov syndróm
Angelmanov syndróm je lekársky názov pre chromozomálnu patológiu, ale zďaleka nie je jediný. Ľudia túto chorobu nazývajú syndrómom bábikových detí, syndrómom šťastnej bábky, Petruškov syndróm a syndrómom smejúcej sa bábiky. Ľudia si vymýšľajú všelijaké názvy (niekedy dokonca urážlivé pre samotných pacientov a ich rodičov), ale choroba je choroba, bez ohľadu na to, aká smiešna môže vyzerať a bez ohľadu na to, aké sú jej dôvody.
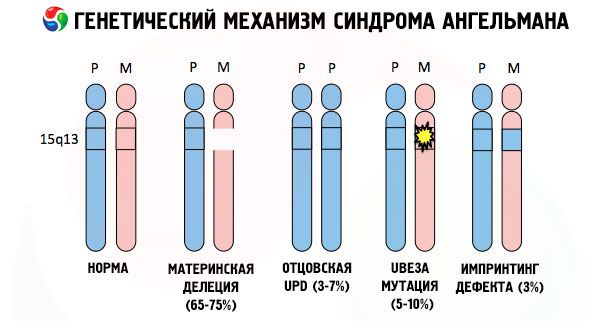
A dôvody vzniku Angelmanovho syndrómu, rovnako ako mnoho iných genetických patológií, sú vo všetkých prípadoch poruchy v štruktúre jedného z chromozómov alebo chromozómovej sady ako celku. Ale v našom prípade celý problém spočíva v chromozóme 15, ktorý sa zdedil od matky. To znamená, že otcovský chromozóm v tomto prípade nemá žiadne odchýlky, ale ženský chromozóm prechádza určitými mutáciami.
Podľa typu chromozomálnej abnormality sa Angelmanov syndróm klasifikuje ako chromozomálna mutácia. Za takéto mutácie sa považujú:
- Delécia (absencia časti chromozómu obsahujúcej určitú sadu génov; ak jeden z génov chýba, hovoríme o mikrodelécii), ktorá je výsledkom dvoch zlomov a jedného spojenia, kedy sa stratí časť pôvodného chromozómu.
- Duplikácia (prítomnosť ďalšej časti chromozómu, ktorá je kópiou existujúcej časti), ktorá vo väčšine prípadov vedie k smrti osoby a menej často k neplodnosti.
- Inverzia (otočenie jednej z častí chromozómu o 180 stupňov, teda opačným smerom, a potom sú v ňom gény umiestnené v opačnom poradí), keď sú prerušené konce chromozómu spojené v poradí inom ako pôvodnom.
- Vloženie (ak je časť genetického materiálu v chromozóme na nesprávnom mieste),
- translokácia (ak je určitá časť chromozómu pripojená k inému chromozómu; takáto mutácia môže byť vzájomná bez straty častí).
Ak dieťa dostane mutovaný chromozóm od nič netušiacej matky, je odsúdené na narodenie s abnormalitami. Za najčastejšiu príčinu Angelmanovho syndrómu sa stále považuje delécia materského 15. chromozómu, keď chýba malá časť. Menej časté mutácie pri syndróme „smejúcej sa bábiky“ sa považujú za:
- translokácia,
- unipaternálna disómia (ak dieťa dostalo od otca pár chromozómov, materský chromozóm chýba),
- mutácia génov v DNA, ktoré sú zároveň hlavným stavebným (genetickým) materiálom a návodom na jeho správne použitie (najmä mutácia génu ube3a v materskom chromozóme).
Prítomnosť jednej z týchto mutácií u rodičov je rizikovým faktorom pre rozvoj Angelmanovho syndrómu u detí. Nielen chromozomálne mutácie, ale aj genomické (ktoré sú spojené s kvantitatívnou zmenou chromozómových súborov a sú častejšie ako chromozomálne) môžu vyvolať rozvoj ochorenia u dieťaťa. Medzi bežné genomické mutácie patrí chromozómová trizómia (ak chromozómová sada osoby má viac ako 46 chromozómov).
Na to, aby sa u dieťaťa objavila patológia, nie je vôbec potrebné, aby rodičia mali chromozomálne abnormality. Napriek tomu existuje určité percento pacientov, ktorých ochorenie je dedičné.
Patogenézy
Poďme sa trochu hlbšie ponoriť do biológie, alebo presnejšie do genetiky. Genetická informácia každého jednotlivého ľudského organizmu je obsiahnutá v 23 pároch chromozómov. Jeden chromozóm z páru sa na dieťa prenáša od otca, druhý od matky. Všetky páry chromozómov sa líšia tvarom a veľkosťou a nesú určité informácie. 23. pár chromozómov (chromozómy X a Y) je teda zodpovedný za formovanie pohlavných znakov dieťaťa (XX - dievča, XY - chlapec, pričom chromozóm Y môže dieťa dostať iba od otca).
V ideálnom prípade dieťa od rodičov dostane 46 chromozómov, ktoré tvoria jeho genetické vlastnosti a predurčujú ho ako jednotlivca. Väčší počet chromozómov sa nazýva trizómia a považuje sa za odchýlku od normy. Napríklad prítomnosť chromozómu 47 v chromozómovej sade (karyotyp, určujúci druh a individuálne vlastnosti) spôsobuje výskyt Downovho syndrómu.
Ak sú chromozómy zafarbené špeciálnym farbivom, potom pod mikroskopom môžete pozdĺž každého z nich vidieť pruhy rôznych odtieňov. Vo vnútri každého pruhu sa nachádza obrovské množstvo génov. Všetky tieto pruhy sú vedcami očíslované a majú pevnú polohu. Absencia jedného z pruhov sa považuje za odchýlku od normy. Pri Angelmanovom syndróme možno veľmi často pozorovať absenciu segmentov materského chromozómu v intervale q11-q13, ktoré sa nachádzajú v dlhom ramene, pričom počet DNA báz je len asi 4 milióny.
Hlavnou zložkou chromozómu je neuveriteľne dlhá molekula DNA obsahujúca tisíce génov a desiatky a stovky miliónov dusíkatých báz. Chromozóm 15, zodpovedný za vývoj Angelmanovho syndrómu a niekoľkých ďalších, teda obsahuje 1200 génov a približne 100 miliónov báz. Akékoľvek narušenie štruktúry molekuly DNA určite ovplyvní vzhľad a vývoj budúceho dieťaťa.
Genetická informácia obsiahnutá v génoch sa premieňa na proteín alebo RNA. Tento proces sa nazýva génová expresia. Týmto spôsobom genetická informácia prijatá od rodičov nadobúda formu aj obsah, ktorý je stelesnený v ich jedinečnom ženskom alebo mužskom dedičovi.
Existuje množstvo patológií s neklasickým typom dedičnosti, vrátane Angelmanovho syndrómu, pri ktorom gény prijaté od rodičov ako súčasť párových chromozómov nesú jedinečný odtlačok rodičov a prejavujú sa rôznymi spôsobmi.
Angelmanov syndróm je teda pozoruhodným príkladom genomického imprintingu, pri ktorom je expresia génov v tele dieťaťa priamo závislá od toho, od ktorého rodiča boli alely prijaté (rôzne formy jedného génu, prijaté od otca a matky, umiestnené na identických úsekoch párových chromozómov). To znamená, že k rozvoju syndrómu vedú iba anomálie v materskom chromozóme, zatiaľ čo mutácie a štrukturálne poruchy otcovského chromozómu spôsobujú úplne odlišné patológie.
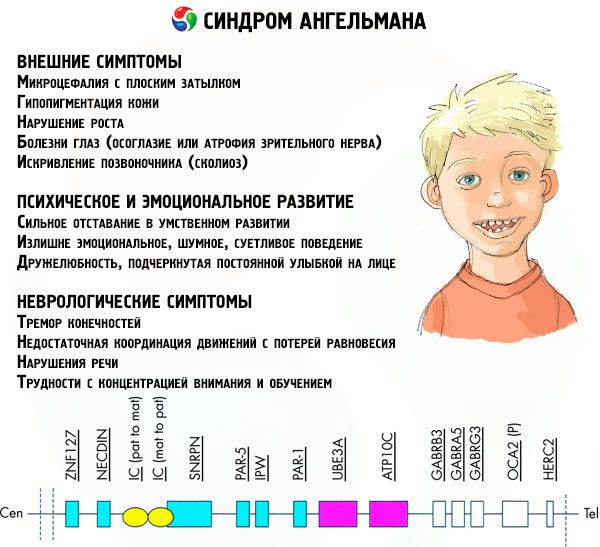
Pri tejto patológii dochádza k nedostatku určitých génov v materskom chromozóme alebo k strate/zníženiu aktivity jednotlivých génov (vo veľkej väčšine prípadov ide o gén ube3a, ktorý sa podieľa na metabolizme ubikvitínu, proteínu regulujúceho degradáciu iných proteínov). V dôsledku toho sú u dieťaťa diagnostikované mentálne vývojové abnormality a fyzické deformácie.
Príznaky Angelmanov syndróm
Príznaky Angelmanovho syndrómu ovplyvňujú rôzne aspekty života a vývoja dieťaťa: fyzické, neurologické, psychické. Na základe toho možno identifikovať 3 skupiny príznakov, ktoré naznačujú vývoj tejto patológie.
- Vonkajšie alebo fyzické príznaky:
- neúmerne malá hlava v porovnaní s telom a končatinami, ktoré majú normálnu veľkosť,
- príliš široké ústa,
- na tvári je takmer vždy úsmev (s otvorenými ústami),
- riedke zuby,
- úzka horná pera,
- často vyplazovaný široký jazyk,
- vyčnievajúca spodná čeľusť,
- špicatá brada,
- veľmi svetlá pokožka, často vlasy (albinizmus, spojený so skutočnosťou, že telo neprodukuje pigment melanín),
- tmavé škvrny na svetlej pokožke (hypopigmentácia v dôsledku nedostatočnej produkcie melanínu)
- fyzické alebo vonkajšie príznaky: očné ochorenia, ako je strabizmus alebo atrofia zrakového nervu,
- zakrivenie chrbtice (skolióza),
- stuhnuté nohy (pri chôdzi si človek neohýba nohy v kolenách kvôli nízkej pohyblivosti kĺbov, preto porovnanie s chôdzou bábiky).
- Príznaky súvisiace s mentálnym a emocionálnym vývojom:
- ťažká mentálna retardácia,
- prehnane emotívne, hlučné a rozrušené správanie,
- časté tlieskanie rukami,
- prejavená priateľskosť, zdôraznená neustálym úsmevom na tvári,
- častý smiech bez dôvodu.
- Neurologické príznaky:
- tremor končatín,
- nedostatočná koordinácia pohybov so stratou rovnováhy,
- znížený svalový tonus,
- rôzne poruchy spánku,
- časté hysterické záchvaty v detstve,
- poruchy reči (dieťa začína rozprávať neskoro, má slabé komunikačné schopnosti a nezrozumiteľnú reč),
- hyperaktivita na pozadí zvýšenej vzrušivosti,
- ťažkosti s koncentráciou a učením.
Ale toto je zovšeobecnený obraz ochorenia. V skutočnosti klinický obraz Angelmanovho syndrómu do značnej miery závisí od štádia vývoja ochorenia a typu chromozomálnej mutácie, ktorá spôsobila patológiu. To znamená, že príznaky ochorenia sa u rôznych pacientov môžu výrazne líšiť, čo nám dlho neumožňovalo odlíšiť patológiu od iných s podobným klinickým obrazom.
Z celkového počtu symptómov môžeme zdôrazniť tie, ktoré sú charakteristické pre všetkých pacientov bez výnimky:
- ťažká mentálna retardácia,
- nevhodné správanie (neprimeraný smiech, zvýšená podráždenosť, slabá koncentrácia, stav eufórie),
- nedostatočný rozvoj motorických zručností,
- zlá koordinácia pohybov, ataxia chôdze (nerovnomerné tempo, kymácanie sa zo strany na stranu atď.), tras končatín.
- porucha vývoja reči s prevahou neverbálnych komunikačných prostriedkov.
Medzi príznakmi, s ktorými sa stretáva prevažná väčšina pacientov, možno rozlíšiť nasledujúce:
- nepomer medzi hlavou a telom spôsobený oneskoreným fyzickým vývojom,
- u mnohých pacientov je tvar lebky taký, že veľkosť mozgu zostáva menšia ako u zdravých ľudí (mikrocefália),
- epileptické záchvaty pred dovŕšením 3 rokov s progresívnym poklesom sily a frekvencie vo vyššom veku,
- skreslenie EEG parametrov (fluktuácie a vysoká amplitúda nízkofrekvenčných vĺn).
Tieto príznaky sú pomerne bežné, avšak 20 % pacientov s Angelmanovým syndrómom ich nemá.
Ešte menej často je možné diagnostikovať také prejavy ochorenia, ako sú:
- ťažký alebo mierny strabizmus,
- slabá kontrola pohybu jazyka, čo má za následok, že pacienti často vyplazujú jazyk bezdôvodne,
- ťažkosti s prehĺtaním a saním, najmä u malých detí,
- narušenie pigmentácie kože a očí,
- zdvihnuté alebo pokrčené ruky pri chôdzi,
- hyperreflexia,
- poruchy spánku, najmä v detstve,
- časté slinenie,
- nenásytný smäd,
- príliš aktívne žuvacie pohyby,
- precitlivenosť na teplo,
- plochá zadná časť hlavy,
- vyčnievajúca spodná čeľusť,
- hladké dlane.
Pomerne veľké percento pacientov má problémy s močením, ktoré zle kontrolujú, zhoršenú jemnú motoriku, čo spôsobuje ťažkosti so starostlivosťou o seba a učením, a nadváhu. Takmer všetci pacienti prežívajú pubertu neskôr ako zdraví rovesníci.
Deti s Angelmanovým syndrómom dobre vnímajú ústnu reč a rozumejú jej, ale nechcú sa zapájať do konverzácie a obmedzujú svoju reč na niekoľko desiatok slov potrebných v každodennom živote. V dospelosti však takíto pacienti vyzerajú mladšie ako ich rovesníci bez genetických patológií.
Mnohé príznaky Angelmanovho syndrómu sú nestále, takže klinický obraz ochorenia sa s vekom výrazne mení. Kŕče a epileptické záchvaty sa stávajú menej častými alebo úplne vymiznú, pacient sa stáva menej vzrušivým a zlepšuje sa spánok.
Komplikácie a následky
Angelmanov syndróm je závažná, v súčasnosti prakticky nevyliečiteľná chromozomálna patológia, ktorá pacientom bráni v možnosti žiť normálny život. Aký bude život dieťaťa s AS, do značnej miery závisí od typu chromozomálnej abnormality.
Duplikácia segmentu chromozómu je vo väčšine prípadov nezlučiteľná so životom. A aj keď takíto pacienti nezomrú v detstve a dosiahnu pubertu, nemajú šancu mať deti.
Delécia alebo absencia časti génov, ktorá sa najčastejšie vyskytuje pri Angelmanovom syndróme, je prekážkou pre dieťa, aby sa naučilo chodiť a rozprávať. Takéto deti majú závažnejšiu formu mentálnej retardácie a epileptické záchvaty sa vyskytujú častejšie a ich intenzita je oveľa väčšia ako u pacientov s inými chromozomálnymi abnormalitami.
Ak ide len o mutáciu jedného génu, s náležitou pozornosťou a prístupom sa dieťa môže naučiť základom starostlivosti o seba, komunikácie a interakcie v skupine, hoci bude vo vývoji stále zaostávať za svojimi rovesníkmi.
Pre deti s Angelmanovým syndrómom, ktoré sú od prírody láskavé, je najdôležitejšia láska a pozornosť rodičov. Len v tomto prípade prinesie vzdelávanie dieťaťa ovocie, aj keď malé. Pacienti s AS samozrejme nebudú môcť študovať v bežnej škole. Potrebujú špeciálne triedy, kde sa deti najprv naučia sústrediť sa a potom sa im postupne budú odovzdávať základy školských vedomostí.
Diagnostika Angelmanov syndróm
Angelmanov syndróm je vrodená vývojová patológia. Za určitých okolností je však často nemožné ho diagnostikovať v dojčenskom a ranom detstve. Je to spôsobené nešpecifickosťou a slabým prejavom symptómov u dojčiat a detí mladších ako 3 roky. A prevalencia ochorenia v našej krajine nie je taká veľká, aby sa lekári naučili rozpoznať ho medzi jej rovesníkmi.
Angelmanov syndróm u dojčiat sa môže prejaviť zníženým svalovým tonusom, ktorý sa prejavuje problémami s kŕmením (slabosť sacieho a prehĺtacieho reflexu) a neskôr ťažkosťami s učením sa chôdze (takéto deti začínajú chodiť oveľa neskôr). Tieto príznaky sú prvými príznakmi vývojovej abnormality u dieťaťa, ktorá môže byť spojená s chromozómovou abnormalitou. Tento predpoklad môže potvrdiť iba genetická analýza.
Zvláštna pozornosť sa venuje deťom, ktorých rodičia majú rôzne genomické alebo chromozomálne poruchy. Ochorenie sa totiž spočiatku nemusí prejaviť a ak sa patológia zistí včas, začatím intenzívnej práce s dieťaťom je možné dosiahnuť výrazne väčší úspech v učení, čím sa spomalí progresia ochorenia.
Ak majú rodičia rôzne chromozomálne abnormality, genetická analýza sa vykonáva ešte pred narodením dieťaťa, pretože SA je jednou z patológií, ktoré možno zistiť v embryonálnom štádiu.
Zber materiálu na genetický výskum sa môže vykonávať dvoma spôsobmi:
- invazívne (s určitým percentom rizika, pretože na odber vzorky plodovej vody je potrebné preniknúť do maternice),
- neinvazívna (analýza DNA dieťaťa z krvi matky).
Potom sa vykonajú nasledujúce štúdie:
- fluorescenčná in situ hybridizácia (metóda FISH) – väzba DNA sondy označenej špeciálnym farbivom na študovanú DNA s následným vyšetrením pod mikroskopom.
- analýza mutácií v géne ube3a a imprintovaných génoch,
- Analýza metylácie DNA pomocou špeciálnych metód používaných v genetike.
Genetické testy poskytujú pomerne presné informácie v prípade chromozómových abnormalít, čo znamená, že budúci rodičia vopred vedia, na čo sa majú pripraviť. Existujú však výnimky. U určitej skupiny pacientov, ak sú prítomné všetky príznaky naznačujúce patológiu, výsledky testov zostávajú normálne. To znamená, že patológiu možno identifikovať iba starostlivým pozorovaním dieťaťa od raného detstva: ako je, kedy začal chodiť a rozprávať, či si pri chôdzi ohýba nohy atď.
Okrem metódy FISH možno medzi inštrumentálnymi diagnostickými metódami Angelmanovho syndrómu rozlíšiť tomografiu (CT alebo MRI), ktorá pomáha určiť stav a veľkosť mozgu, a elektroencefalogram (EEG), ktorý ukazuje, ako fungujú jednotlivé časti mozgu.
Lekári zvyčajne stanovia konečnú diagnózu vo veku 3-7 rokov, keď už má pacient väčšinu príznakov a je viditeľná dynamika vývoja ochorenia.
Aké testy sú potrebné?
Odlišná diagnóza
Angelmanov syndróm je genetická patológia, ktorá prakticky nemá žiadne špecifické prejavy. Väčšina symptómov môže rovnako naznačovať AS aj iné genetické patológie.
Diferenciálna diagnostika Angelmanovho syndrómu sa vykonáva s nasledujúcimi patológiami:
- Pitt-Hopkinsov syndróm (pacienti sa vyznačujú mentálnou retardáciou, veselou povahou, úsmevom, majú pomerne veľké a široké ústa, je zaznamenaná mikrocefália). Rozdielom sú záchvaty hyperventilácie a zadržiavanie dychu v bdelom stave.
- Christiansonov syndróm (pacienti sú mentálne retardovaní ľudia s veselou povahou, neschopní hovoriť, charakterizovaní mikrocefaliou, ataxiou, kŕčmi, mimovoľnými pohybmi svalov).
- Mowat-Wilsonov syndróm (príznaky: mentálna retardácia, epileptické záchvaty, špicatá brada, otvorené ústa, šťastný výraz tváre, mikrocefália). Rozlišovací znak: veľká vzdialenosť medzi očami, oči sklonené dovnútra, zaoblená špička nosa, dozadu otočená ušnica.
- Kabukiho syndróm (charakterizovaný miernou až stredne ťažkou mentálnou retardáciou, rečovými a motorickými problémami, svalovou slabosťou, epileptickými záchvatmi, mikrocefáliou, dlhými intervalmi medzi svrbením a zhoršenou koordináciou). Charakterizovaný klenutým obočím, vytočenou laterálnou časťou dolného viečka, široko posadenými očami, dlhými očnými štrbinami s dlhými, hustými mihalnicami.
- Rettov syndróm (odlišovanie od AS u žien). Príznaky: oneskorený vývoj reči, záchvaty, mikrocefália. Rozdiel je v tom, že na tvári nie je šťastný výraz, vyskytujú sa záchvaty apnoe a apraxie, ktoré časom progredujú.
- Autozomálne recesívny syndróm mentálnej tardácie 38 (príznaky: výrazná mentálna retardácia s oneskorením motorických zručností a reči, svalová slabosť, problémy s kŕmením v detstve, impulzivita). Charakteristickým znakom je modrá farba dúhovky.
- Syndróm duplikácie génu MECP 2 (odlišuje sa od SA u mužov). Príznaky: ťažká mentálna retardácia, svalová slabosť od detstva, problémy s rečou alebo jej absencia, epilepsia. Rozlišovacie znaky: progresívna myopatia, neustále sa opakujúce infekcie.
- Kleefstrov syndróm (príznaky: problémy s rečou a myslením, svalová slabosť, poruchy spánku, nedostatok pozornosti, otvorené ústa, hyperaktivita, záchvaty, ataxia, poruchy rovnováhy). Charakteristické znaky: plochá tvár, krátky tupý nos, široko posadené oči, veľká vytočená spodná pera, agresívne výbuchy.
- Smithov-Magenisov syndróm (charakterizovaný záchvatmi, problémami so spánkom, poruchami intelektuálneho a motorického vývoja). Medzi charakteristické znaky patrí široká a plochá tvár a výrazné čelo.
- Koolen-de Vriesov syndróm (mierna až stredne ťažká mentálna retardácia, svalová slabosť, záchvaty, priateľskosť). Charakteristické znaky: dlhá tvár s vysokým čelom, odstávajúce uši, šikmé oči, vysoká pohyblivosť kĺbov, vrodené srdcové chyby.
- Phelanov-McDermidov syndróm (príznaky: mentálna retardácia, poruchy reči alebo nedostatok reči). Rozlišovacie znaky: veľké ruky s vyvinutými svalmi, svalová slabosť od narodenia, slabé potenie.
Takéto patológie, ako je deficit adenylsukcinátu, syndróm autozomálne recesívnej mentálnej retardácie 1, syndróm duplikácie chromozómu 2q23.1, syndrómy haploinsuficiencie génov FOXG1, STXBP1 alebo MEF2C a niektoré ďalšie, sa môžu „chváliť“ príznakmi podobnými Angelmanovmu syndrómu.
Úlohou lekára je stanoviť presnú diagnózu, odlíšiť Angelmanov syndróm od patológií s podobnými príznakmi a predpísať účinnú liečbu, ktorá je relevantná pre diagnostikované štádium ochorenia.
Liečba Angelmanov syndróm
Angelmanov syndróm je jednou z tých patológií, pre ktoré medicína stále hľadá účinnú liečbu. Etiologická liečba ochorenia je vo fáze vývoja rôznych metód a prostriedkov, z ktorých mnohé ešte neboli testované na ľuďoch. To znamená, že lekári sa zatiaľ musia obmedziť na symptomatickú liečbu, ktorá pomáha nejakým spôsobom zmierniť nezávideniahodnú situáciu detí a dospelých s marionetovým syndrómom, trpiacich epileptickými záchvatmi, slinením, hypotenziou a poruchami spánku.
Pomocou správne zvoleného antikonvulzívneho lieku je teda možné znížiť frekvenciu a silu epileptických záchvatov. Celý problém však spočíva v tom, že záchvaty u pacientov so SA sa od bežných epileptických záchvatov líšia tým, že sa vyznačujú niekoľkými typmi záchvatov, čo znamená, že stav možno zmierniť podávaním viacerých liekov naraz.
Najobľúbenejšie antikonvulzíva používané na liečbu Angelmanovho syndrómu sú: kyselina valproová, topiramát, lamotrigín, levetiracetam, klonazepam a lieky na ich báze. Menej často sa používajú lieky na báze karmazepínu, fenytoínu, fenobarbitalu, etosuximidu, pretože niektoré z nich môžu vyvolať paradoxný účinok spočívajúci v zosilnení a zvýšení frekvencie epileptických záchvatov. Toto sa stane, ak sa liek používa ako súčasť monoterapie.
Na liečbu slintania sa zvyčajne používajú dve metódy: medikamentózna (lieky, ktoré potláčajú tvorbu slín) a chirurgická, ktorá zahŕňa reimplantáciu slinných kanálikov. V prípade sankčnej atrofie sa však tieto metódy považujú za neúčinné a otázka zostáva otvorená. Rodičia a tí, ktorí sa o takýchto pacientov starajú, musia tejto problematike venovať osobitnú pozornosť, pretože samotní pacienti zvyčajne nekontrolujú slinenie a niektorí sa o seba jednoducho nedokážu postarať.
Ďalším problémom je krátka dĺžka spánku. Deti s Angelmanovým syndrómom často spia maximálne 5 hodín, čo má negatívny vplyv na fungovanie celého tela. Ľahko vzrušivé, aktívne deti, ktoré milujú hry a komunikáciu (aj keď sa snažia obmedziť na neverbálne metódy), sú počas dňa citeľne unavené. Aby si telo dobre oddýchlo, potrebuje hlboký a plnohodnotný spánok, ale práve v tom je háčik.
Zdá sa, že sedatívne lieky (fenotiazíny a atypické antipsychotiká), ktoré upokojujú nervový systém, by mali byť postačujúce na zlepšenie spánku u excitovaných pacientov. V prípade AS je však užívanie takýchto liekov spojené s výskytom negatívnych účinkov. Preto lekári stále uprednostňujú mierne lieky na spanie, ako je melatonín (prírodný hormonálny liek založený na spánkovom hormóne), ktorý sa pacientom podáva hodinu pred spaním v množstve 1 tablety, a difenhydramín. Frekvenciu podávania a dávkovanie určuje lekár v závislosti od stavu a veku pacienta.
Pacienti s Angelmanovým syndrómom majú niekedy problémy s trávením a stolicou. Stolicu môžete zlepšiť preháňadlami (najlepšie bylinnými).
Alebo môžete k problému pristupovať inak, ako to urobili americkí lekári, na základe niektorých metód liečby autizmu, pretože mnohé príznaky charakteristické pre AS sú charakteristické aj pre autizmus (impulzivita, mimovoľné pohyby, opakujúce sa činnosti, deficit pozornosti, komunikačné problémy atď.). Bolo zaznamenané, že zavedenie hormónu sekretínu, ktorý normalizuje trávenie a stolicu, má pozitívny vplyv na pozornosť pacientov a oxytocín pomáha zlepšovať kognitívne schopnosti a pamäť dieťaťa a korigovať správanie.
Pravda, samotné hormóny nestačia, najmä pokiaľ ide o deti. Pri Angelmanovom syndróme je indikovaná behaviorálna terapia, práca s psychológom a logopédom (výučba neverbálnych komunikačných metód a posunkovej reči). Vzdelávanie takýchto detí by malo byť založené na individuálnom programe za účasti špeciálne vyškolených učiteľov, psychológa a rodičov. Bohužiaľ, nie všade je to možné a rodiny zostávajú so svojím problémom samé.
Keďže mnoho mladých pacientov s AS trpí nízkym svalovým tonusom a problémami s kĺbmi, veľká pozornosť sa venuje fyzioterapii. Najčastejšie sa lekári uchyľujú k parafínovým aplikáciám, elektroforéze a magnetoterapii.
Aktívna tonizujúca masáž a špeciálne cvičenia liečebného telesného tréningu pomôžu chorému dieťaťu po čase postaviť sa na nohy a s istotou chodiť. V tomto smere je obzvlášť užitočná aquagymnastika, ktorá sa odporúča pri SA v studenej vode. Zvyšuje svalový tonus a učí dieťa ovládať svoje telo a koordinovať pohyby.
Antikonvulzívna liečba
Najnebezpečnejším príznakom Angelmanovho syndrómu sú záchvaty podobné epilepsii. Tento príznak sa pozoruje u 80 % pacientov, čo znamená, že všetkým je potrebné predpísať účinnú antikonvulzívnu liečbu.
Liečba epileptických záchvatov sa vykonáva pomocou vitamínov a antikonvulzív. Pri Angelmanovom syndróme, sprevádzanom konvulzívnym syndrómom, budú užitočné vitamíny skupiny B, ako aj vitamíny C, D a E. Samostatné predpisovanie vitamínovej terapie je však v tomto prípade veľmi nebezpečné, pretože nekontrolovaný príjem vitamínov môže znížiť účinnosť antiepileptík a vyvolať nové, závažnejšie a dlhšie trvajúce záchvaty.
Výber antikonvulzívnych liekov a predpísanie ich účinného dávkovania by mal tiež vykonať špecializovaný lekár. Ten tiež rozhodne, či bude stačiť jeden liek, alebo či bude pacient musieť dlhodobo užívať 2 alebo viac liekov.
Väčšine pacientov lekári predpisujú lieky s kyselinou valproovou (kyselina valproová, Depakine, Convulex, Valparin atď.), ktoré zabraňujú záchvatom a zlepšujú náladu a duševný stav pacientov.
Kyselina valproová je dostupná vo forme tabliet, sirupu a injekčných roztokov. Najobľúbenejším liekom je liek s predĺženým uvoľňovaním "Depakine" v tabletách a ako roztok na intravenózne podanie. Dávkovanie lieku určuje lekár individuálne v závislosti od hmotnosti, veku a stavu pacienta.
Liek sa užíva počas jedla 2 až 3-krát denne. Priemerná denná dávka je 20 – 30 mg na 1 kilogram hmotnosti pacienta, maximálna je 50 mg/kg denne.
Kontraindikácie pre použitie. Nepoužívajte v prípade dysfunkcie pečene a pankreasu, hemoragickej diatézy, hepatitídy, porfýrie a precitlivenosti na liek.
Medzi vedľajšie účinky patrí trasenie rúk, poruchy trávenia a stolice a zmeny telesnej hmotnosti.
"Topiramát" je tiež liekom voľby pre SA. Vyrába sa vo forme tabliet a používa sa ako súčasť monoterapie, tak aj v kombinácii s inými liekmi.
Spôsob podávania a dávkovanie. Tablety sa užívajú perorálne bez ohľadu na príjem jedla. Počiatočná denná dávka pre dospelých je 25 – 50 mg, pre deti – 0,5 – 1 mg/kg. Každý týždeň sa dávka zvyšuje podľa pokynov lekára.
Liek sa nemá užívať počas tehotenstva a laktácie, ako aj v prípade precitlivenosti na jeho zložky. Liek má mnoho rôznych vedľajších účinkov.
Lieky, ktoré môže lekár predpísať na Angelmanov syndróm: Clomazepam, Rivotril, Lamotrigín, Seizar, Lamictal, Levetiracetam, Keppra, Epiterra atď.
 [ 16 ], [ 17 ], [ 18 ], [ 19 ]
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Tradičná medicína a homeopatia
Tradičná medicína, rovnako ako homeopatické prípravky, je samozrejme relatívne bezpečná, ale účinnosť takejto liečby Angelmanovho syndrómu možno považovať za kontroverznú.
Hoci ľudová liečba môže v niektorých veciach stále pomôcť. Hovoríme o zastavení epileptických záchvatov. V tomto ohľade môže byť liečba bylinkami celkom účinná.
Dobrý účinok poskytuje liečivá kolekcia na báze pivonky, sladkého drievka a žaburinky (zložky sa užívajú v rovnakom množstve). Byliny je potrebné rozomlieť na múku. Po 2 týždňoch od začiatku užívania si môžete všimnúť výrazný pokles frekvencie záchvatov.
Pri kŕčoch je užitočný aj odvar z levandule (1 čajová lyžička na pohár vriacej vody). Zmes sa varí 5 minút a lúhuje sa pol hodiny. Liek sa užíva v noci počas 14 dní.
Vodný (alebo alkoholový) nálev z materskej dúšky sa považuje za účinný pri epileptických záchvatoch.
Z homeopatických prípravkov na prevenciu záchvatov pri Angelmanovom syndróme môžete použiť lieky na báze harmančeka a materskej dúšky, Acidum hydrocyanicum, Argentum nitricum, Kalium bromatum, Arsenicum album. Treba však vziať do úvahy, že účinné a bezpečné dávky liekov môže v každom konkrétnom prípade predpísať iba homeopatický lekár.
Prevencia
Ako čitateľ pravdepodobne už pochopil, medicína zatiaľ nedokáže zabrániť génovým mutáciám a iným chromozomálnym abnormalitám, ani situáciu napraviť. Môže sa to stať každému, pretože deti s Angelmanovým syndrómom sa rodia zdravým rodičom a genetika, ktorá je v súčasnosti jednou z najmenej študovaných oblastí medicíny, to zatiaľ nedokáže vysvetliť.
Jediné, čo sa dá urobiť, je zodpovedne pristupovať k plánovaniu tehotenstva, včas sa zaregistrovať a podstúpiť vyšetrenia. Ale opäť, takéto opatrenie bude skôr vzdelávacie ako preventívne, ako akékoľvek vyšetrenia. Mladí rodičia však budú vopred vedieť, na čo sa majú pripraviť, a v prípade kladnej odpovede sa rozhodnú, či si môžu vziať na seba takú zodpovednosť, ako je výchova chorého dieťaťa.
Predpoveď
Prognóza Angelmanovho syndrómu závisí od povahy chromozómovej abnormality a včasnosti jej detekcie. Najviac sú postihnuté tie deti, ktorých chromozóm 15 obsahuje „medzery“ v génoch (delécia). Pravdepodobnosť, že takíto pacienti budú chodiť a rozprávať, je extrémne nízka. Iné prípady sa dajú napraviť starostlivým prístupom a láskou k vášmu dieťaťu.
Bohužiaľ, takíto pacienti sa nebudú môcť stať plnohodnotnými členmi spoločnosti, napriek tomu, že zďaleka nie sú hlúpi, rozumejú reči a jej významu. S komunikáciou však budú mať problémy po zvyšok života. Pacientov možno učiť posunkovej reči od detstva, ale nemožno ich nútiť komunikovať pomocou slov. Slovná zásoba „hovoriacich“ pacientov je obmedzená na minimum slov používaných v každodennom živote (5 – 15 slov).
Čo sa týka dĺžky života a celkového zdravotného stavu pacientov s Angelmanovým syndrómom, čísla sa tu pohybujú okolo priemerných hodnôt. V dospelosti pacienti väčšinou čelia zdravotným problémom, ako je skolióza a obezita, ktoré pri správnom prístupe k liečbe neohrozujú život.