Lekársky expert článku

Nové publikácie
Mukopolysacharidóza typu 3
Posledná kontrola: 04.07.2025

Všetok obsah iLive je lekársky kontrolovaný alebo kontrolovaný, aby sa zabezpečila čo najväčšia presnosť faktov.
Máme prísne smernice týkajúce sa získavania zdrojov a len odkaz na seriózne mediálne stránky, akademické výskumné inštitúcie a vždy, keď je to možné, na lekársky partnerské štúdie. Všimnite si, že čísla v zátvorkách ([1], [2] atď.) Sú odkazmi na kliknutia na tieto štúdie.
Ak máte pocit, že niektorý z našich obsahov je nepresný, neaktuálny alebo inak sporný, vyberte ho a stlačte kláves Ctrl + Enter.
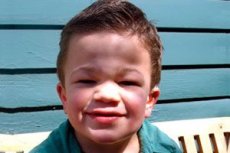
Mukopolysacharidóza typu III (synonymá: Sanfilippov syndróm, lyzozomálny deficit aN-acetylglukozaminidázy - mukopolysacharidóza III A, deficit acetyl-CoA-a-glukozaminid-N-acetyltransferázy - mukopolysacharidóza III B, N-acetylglukozamín-6-sulfatáza - mukopolysacharidóza III C, deficit sulfamidázy - mukopolysacharidóza III D).
 [
[ Patogenézy
Ochorenie je spôsobené mutáciami v štyroch rôznych génoch: lyzozomálna α-acetylglukozaminidáza (mukopolysacharidóza III A), acetyl-CoA-α-glukozaminid-N-acetyltransferáza (mukopolysacharidóza III B), lyzozomálna N-acetylglukozamín-6-sulfatáza (mukopolysacharidóza III C) a sulfamidáza (mukopolysacharidóza III D). Všetky enzýmy sa podieľajú na metabolizme heparánsulfátu.
Gén heparan-N-sulfatázy – SGSH – sa nachádza na dlhom ramene chromozómu 17 – 17q25.3. 75,3 % v súčasnosti známych mutácií v géne SGSH sú bodové mutácie. Boli opísané často sa vyskytujúce mutácie charakteristické pre európske populácie – R74C (56 % v Poľsku a 21 % v Nemecku) a R245H (56 % v Holandsku).
Frekvencia mutácie R74C je 47,5 %, mutácie R245H je 7,5 %. Ďalšie dve opísané mutácie, delll35G a N389S, spolu predstavujú 21,7 % mutantných alel.
Gén pre aN-acetyl-glukozaminidázu (NAGLU) sa nachádza na dlhom ramene chromozómu 17 - 17q21. 69 % mutácií nájdených v géne NAGLU sú missense a nonsense mutácie, 26,3 % sú malé delécie a inzercie. Gén pre acetyl-CoA-cc-glukozaminid-N-acetyltransferázu (HGSNAT) sa nachádza na krátkom ramene chromozómu 8 - 8p11.1. Gén bol charakterizovaný až v roku 2006 a doteraz sa v ňom našlo len niekoľko mutácií.
Gén N-acetyl-glukozamín-6-sulfatázy - GNS - sa nachádza na dlhom ramene chromozómu 12 - 12ql4. Na svete je registrovaných 12 pacientov s mukopolysacharidózou IIID. Boli opísané štyri mutácie v géne GNS.
Pri všetkých podtypoch mukopolysacharidózy III dochádza k poruche degradácie heparánsulfátu, ktorý je súčasťou štruktúry bunkových membrán vrátane neuronálnych membrán, čo koreluje s ťažkým neurodegeneratívnym procesom spôsobeným kortikálnou atrofiou. Chronická hnačka sa vysvetľuje zapojením autonómneho nervového systému do patologického procesu spolu s dysfunkciou črevnej sliznice. Senzorineurálna strata sluchu je pravdepodobne spôsobená tromi príčinami: častými zápalmi stredného ucha, deformáciou sluchových kostičiek a anomáliami vnútorného ucha. Stuhnutosť kĺbov je výsledkom deformácie metafýz, zhrubnutie kĺbového puzdra je sekundárne k ukladaniu glykozaminoglykánov a fibróze. Vnútrosyndromické rozdiely v závažnosti ochorenia sú spôsobené výlučne reziduálnou funkčnou aktivitou mutantného enzýmu: čím je vyššia, tým je ochorenie miernejšie.
Príznaky mukopolysacharidóza typu 3
Klinický polymorfizmus pri Sanfilippovom syndróme je menej výrazný ako pri iných typoch mukopolysacharidózy. Charakteristický je pomalý postup ochorenia, závažné neurologické poruchy s miernymi príznakmi z vnútorných orgánov a iných systémov.
Prvé príznaky ochorenia sa zvyčajne objavujú medzi 2. a 6. rokom života u detí s predtým normálnym vývojom. Medzi manifestné príznaky patrí regresia psychomotorického a rečového vývoja, psychiatrické poruchy vo forme syndrómu hyperaktivity, autistické alebo agresívne správanie, poruchy spánku; deti sa stávajú neopatrnými a nepozornými.
Ďalšími bežnými príznakmi sú hirsutizmus, hrubé ochlpenie, mierna hepatosplenomegália, valgózna deformácia končatín a krátky krk. Vývoj hrubých čŕt tváre, ako je gargoylizmus, a skeletálnych deformít, ako je mnohopočetná dysostóza, je pri mukopolysacharidóze III slabo prejavený v porovnaní s inými typmi mukopolysacharidózy charakterizovanými Hurlerovým fenotypom. Výška spravidla zodpovedá veku a stuhnutosť kĺbov zriedkavo spôsobuje dysfunkciu. U väčšiny pacientov sa často vyvinie osteoporóza a osteomalácia. Sekundárne skeletálne poruchy - vysoké riziko patologických zlomenín. Závažné psychoneurologické poruchy sa najčastejšie pozorujú v 6. až 10. roku života a vedú k výraznej sociálnej maladaptácii. Progresívna senzorineurálna strata sluchu je neoddeliteľnou súčasťou všetkých pacientov s ťažkými a stredne ťažkými formami ochorenia. Kŕče sa pozorujú takmer u všetkých pacientov s progresiou ochorenia.
Ochorenie postupuje rýchlo a väčšina pacientov sa nedožije veku 20 rokov. Mukopolysacharidóza IIIA sa považuje za najbežnejší a najzávažnejší typ tohto syndrómu.
Diagnostika mukopolysacharidóza typu 3
Diagnóza mukopolysacharidózy III sa potvrdzuje stanovením hladiny vylučovania glykozaminoglykánov močom a meraním aktivity enzýmov. V prípade mukopolysacharidózy III sa zvyšuje celkové vylučovanie glykozaminoglykánov močom a pozoruje sa hyperexkrécia heparánsulfátu. Aktivita lyzozomálnych enzýmov zodpovedajúcich určitému podtypu mukopolysacharidózy III sa meria v leukocytoch alebo kultúre kožných fibroblastov pomocou umelého fluorogénneho substrátu.
Prenatálna diagnostika je možná meraním enzýmovej aktivity v biopsii choriových klkov v 9. – 11. týždni tehotenstva a/alebo stanovením spektra glykozaminoglykánov v plodovej vode v 20. – 22. týždni tehotenstva. U rodín so známym genotypom je možné vykonať DNA diagnostiku v ranom štádiu tehotenstva.
Aké testy sú potrebné?
Odlišná diagnóza
Diferenciálna diagnostika sa vykonáva v rámci skupiny mukopolysacharidóz aj s inými lyzozomálnymi ochoreniami: mukolipidózy, galaktosialidóza, sialidóza, manozidóza, fukozidóza, gangliozidóza GM1.
Komu sa chcete obrátiť?
Liečba mukopolysacharidóza typu 3
Doteraz neboli vyvinuté účinné metódy liečby mukopolysacharidózy III. Indikovaná je symptomatická liečba.
Использованная литература