Lekársky expert článku

Nové publikácie
Subakútna nekrotizujúca Leahova encefalomyopatia
Posledná kontrola: 04.07.2025

Všetok obsah iLive je lekársky kontrolovaný alebo kontrolovaný, aby sa zabezpečila čo najväčšia presnosť faktov.
Máme prísne smernice týkajúce sa získavania zdrojov a len odkaz na seriózne mediálne stránky, akademické výskumné inštitúcie a vždy, keď je to možné, na lekársky partnerské štúdie. Všimnite si, že čísla v zátvorkách ([1], [2] atď.) Sú odkazmi na kliknutia na tieto štúdie.
Ak máte pocit, že niektorý z našich obsahov je nepresný, neaktuálny alebo inak sporný, vyberte ho a stlačte kláves Ctrl + Enter.
 [
[ Príčiny Leahovho syndrómu
Základom ochorenia je nedostatok enzýmov, ktoré zabezpečujú produkciu energie, najmä v dôsledku narušenia metabolizmu kyseliny pyruvovej a poruchy transportu elektrónov v dýchacom reťazci. Vyvíja sa deficit komplexu pyruvátdehydrogenázy (podjednotka a-E1), pyruvátkarboxylázy, komplexu 1 (NAD-koenzým Q-reduktáza) a komplexu 4 (cytochrómoxidáza) dýchacieho reťazca.
Bolo zistené, že defekty pyruvátkarboxylázy, komplexu 1 (NAD-koenzým Q-reduktáza) a komplexu 4 (cytochróm oxidáza) dýchacieho reťazca sa dedia autozomálne recesívne, defekty komplexu pyruvátdehydrogenázy (podjednotka a-E1) sa dedia recesívne viazaným spôsobom na chromozóm X. V prípade bodových mutácií mtDNA, ktoré postihujú 6. podjednotku ATPázy, je typická mitochondriálna dedičnosť. Najčastejšie sa vyskytuje miscens mutácia spojená s nahradením tymínu guanínom alebo cytozínom v pozícii 8993 mtDNA. Menej častá je mutácia v pozícii 9176 mtDNA. Vzhľadom na to, že mutácia T8993G je hlavnou poruchou pri NARP syndróme, boli opísané rodiny s týmito dvoma ochoreniami. U detí bola opísaná aj mutácia v mtDNA v pozícii 8344, ktorá sa vyskytuje pri MERRF syndróme.
Predpokladá sa, že v prípade akumulácie mutantnej mtDNA vo väčšine mitochondrií sa vyvinie závažný priebeh Leighovho syndrómu. Pri mitochondriálnej genéze tohto stavu sa mutantná mtDNA nachádza v 90 % všetkých mitochondrií. Patogenéza je spojená s porušením tvorby energie v bunkách a rozvojom laktátovej acidózy.
Príznaky Leahovho syndrómu
Prvé príznaky ochorenia sa objavujú v ranom veku (1-3 roky). Sú však známe prípady manifestácie ochorenia už v 2 týždňoch a v 6-7 rokoch. Najprv sa vyvíjajú nešpecifické poruchy: oneskorený psychomotorický vývoj, znížená chuť do jedla, epizódy vracania, deficit telesnej hmotnosti. Následne sa zhoršujú neurologické príznaky: svalová hypotónia alebo dystónia s prechodom do hypertónie, ataky myoklonu alebo tonicko-klonických záchvatov, tremor končatín, choreoatetóza, poruchy koordinácie, znížené šľachové reflexy, letargia, ospalosť. Mozgová neurodegenerácia je progresívna. Zvyšujú sa príznaky pyramídovej a extrapyramídovej insuficiencie, zhoršuje sa prehĺtanie. Často sa pozorujú zmeny v orgáne videnia, ako je ptóza, oftalmoplégia, atrofia zrakových nervov, menej často pigmentová degenerácia sietnice. Niekedy sa vyvíja hypertrofická kardiomyopatia, objavujú sa epizódy tachypnoe.
Zriedkavo ochorenie prebieha ako akútna encefalopatia. Typickejší je chronický alebo subakútny priebeh, ktorý vedie k fatálnemu koncu niekoľko rokov po nástupe ochorenia. Pri rýchlom priebehu (niekoľko týždňov) nastáva smrť v dôsledku paralýzy dýchacieho centra.
Diagnostika Leahovho syndrómu
Biochemický krvný test odhalí laktátovú acidózu v dôsledku hromadenia kyseliny mliečnej a pyrohroznovej v krvi a mozgovomiechovom moku, ako aj zvýšenie obsahu alanínu v krvi. Môže byť tiež zvýšená hladina ketónových teliesok. V moči sa zistí zvýšené vylučovanie organických kyselín: mliečnej, fumarovej atď. Hladina karnitínu v krvi a tkanivách často klesá.
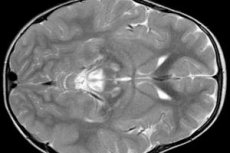
Výsledky EEG odhaľujú fokálne známky epileptickej aktivity. Údaje z MRI odhaľujú zväčšenie mozgových komôr, bilaterálne poškodenie mozgu, kalcifikáciu bazálnych ganglií (nucleus caudatus, putamen, substantia nigra, globus pallidus). Môže sa zistiť aj atrofia mozgových hemisfér a mozgovej hmoty.
Morfologické vyšetrenie odhaľuje hrubé zmeny v mozgovej hmote: symetrické ložiská nekrózy, demyelinizácie a špongióznej degenerácie mozgu, najmä stredných častí, mostíka, bazálnych ganglií, talamu a zrakového nervu. Histologický obraz zahŕňa cystickú degeneráciu mozgového tkaniva, astrocytickú gliózu, neuronálnu smrť a zvýšenie počtu mitochondrií v bunkách. V kostrovom svalstve dochádza k akumulácii lipidových inklúzií, zníženiu histochemickej reakcie na komplexy 1 a 4 dýchacieho reťazca, subsarkolemálnej akumulácii mitochondrií, abnormálnym mitochondriám s dezorganizáciou krist. Fenomén RRF sa často nezistil.
Ako preskúmať?
Aké testy sú potrebné?
Использованная литература