Lekársky expert článku

Nové publikácie
Cystická fibróza
Posledná kontrola: 04.07.2025

Všetok obsah iLive je lekársky kontrolovaný alebo kontrolovaný, aby sa zabezpečila čo najväčšia presnosť faktov.
Máme prísne smernice týkajúce sa získavania zdrojov a len odkaz na seriózne mediálne stránky, akademické výskumné inštitúcie a vždy, keď je to možné, na lekársky partnerské štúdie. Všimnite si, že čísla v zátvorkách ([1], [2] atď.) Sú odkazmi na kliknutia na tieto štúdie.
Ak máte pocit, že niektorý z našich obsahov je nepresný, neaktuálny alebo inak sporný, vyberte ho a stlačte kláves Ctrl + Enter.
Cystická fibróza je genetické autozomálne recesívne monogénne ochorenie charakterizované poruchou sekrécie exokrinných žliaz životne dôležitých orgánov s poškodením predovšetkým dýchacieho a tráviaceho systému, ťažkým priebehom a nepriaznivou prognózou.
 [ 1 ]
[ 1 ]
Epidemiológia
Výskyt cystickej fibrózy kolíše medzi 1:2 500 a 1:4 600 novorodencov. Každý rok sa na celom svete narodí približne 45 000 ľudí s cystickou fibrózou. Výskyt nositeľov génu cystickej fibrózy je 3 – 4 %, pričom na celom svete je nositeľmi tohto génu približne 275 miliónov ľudí, z ktorých približne 5 miliónov žije v Rusku a približne 12,5 milióna v krajinách SNŠ.
Príčiny cystická fibróza
Cystická fibróza sa prenáša autozomálne recesívne. Gén cystickej fibrózy sa nachádza v autozóme 7, obsahuje 27 exónov a pozostáva z 250 000 párov nukleotidov.
Jeden gén môže mať mnoho mutácií, z ktorých každá je špecifická pre konkrétnu populáciu alebo geografickú oblasť. Bolo opísaných viac ako 520 mutácií, z ktorých najbežnejšia je delta-P-508, t. j. substitúcia aminokyseliny fenylalanínu v pozícii 508.
Patogenézy
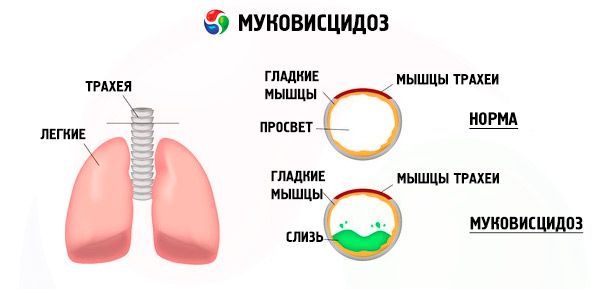
Mutácie v géne cystickej fibrózy narúšajú štruktúru a funkciu proteínu nazývaného CFTR (transmembránový regulátor cystickej fibrózy). Tento proteín funguje ako chloridový kanál zapojený do výmeny vody a elektrolytov v epitelových bunkách bronchopulmonálneho systému, gastrointestinálneho traktu, pankreasu, pečene a reprodukčného systému. V dôsledku narušenia funkcie a štruktúry proteínu CFTR sa vo vnútri bunky hromadia chloridové ióny Cl⁻ . To vedie k zmene elektrického potenciálu v lúmene vylučovacích kanálikov, čo uľahčuje tok veľkého množstva sodných iónov (Na⁺ ) z lúmenu kanálika do bunky a ďalej zvyšuje absorpciu vody z pericelulárneho priestoru.
V dôsledku týchto zmien sa sekrécia väčšiny exokrinných žliaz zahusťuje, jej evakuácia je narušená, čo vedie k výrazným sekundárnym poruchám v orgánoch a systémoch, najvýraznejším v bronchopulmonálnom a tráviacom systéme.
V prieduškách sa vyvíja chronický zápalový proces rôznej intenzity, funkcia ciliárneho epitelu je prudko narušená, spútum sa stáva veľmi viskóznym, hustým, veľmi ťažko sa vyprázdňuje, pozoruje sa jeho stagnácia, tvoria sa bronchiolo- a bronchiektázie, ktoré sa časom vyskytujú častejšie. Tieto zmeny vedú k zvýšeniu hypoxie a vzniku chronického pľúcneho srdcového ochorenia.
Pacienti s cystickou fibrózou sú mimoriadne predisponovaní k rozvoju chronického zápalu v bronchopulmonálnom systéme. Je to spôsobené výraznými poruchami lokálneho bronchopulmonálneho obranného systému (znížené hladiny IgA, interferónu, fagocytárna funkcia alveolárnych makrofágov a leukocytov).
Alveolárne makrofágy hrajú hlavnú úlohu vo vývoji chronického zápalu v bronchopulmonálnom systéme. Produkujú veľké množstvo IL-8, čo dramaticky zvyšuje chemotaxiu neutrofilov v bronchiálnom strome. Neutrofily sa hromadia vo veľkých množstvách v prieduškách a spolu s epitelovými bunkami vylučujú mnoho prozápalových cytokínov vrátane IL-1, 8, 6, faktora nekrózy nádorov a leukotriénov.
Dôležitú úlohu v patogenéze poškodenia bronchopulmonálneho systému zohráva aj vysoká aktivita enzýmu elastázy. Rozlišuje sa exogénna a endogénna elastáza. Prvú produkuje bakteriálna flóra (najmä Pseudomonas aeruginosa), druhú neutrofilné leukocyty. Elastáza ničí epitel a ďalšie štrukturálne prvky priedušiek, čo prispieva k ďalšiemu narušeniu mukociliárneho transportu a rýchlej tvorbe bronchiektázií.
Neutrofilné leukocyty tiež vylučujú ďalšie proteolytické enzýmy. Alfa-1-antipyrzín a sekrečný inhibítor leukoproteáz pôsobia proti vplyvu proteolytických enzýmov, a tým chránia bronchopulmonálny systém pred ich škodlivým vplyvom. U pacientov s cystickou fibrózou sú však tieto ochranné faktory, žiaľ, potlačené významným množstvom neutrofilnej proteázy.
Všetky tieto okolnosti prispievajú k zavedeniu infekcie do bronchopulmonálneho systému a rozvoju chronickej hnisavej bronchitídy. Okrem toho treba vziať do úvahy, že defektný proteín kódovaný génom cystickej fibrózy mení funkčný stav bronchiálneho epitelu, čo podporuje adhéziu baktérií, predovšetkým Pseudomonas aeruginosa, na bronchiálny epitel.
Spolu s patológiou bronchopulmonálneho systému cystická fibróza spôsobuje aj vážne poškodenie pankreasu, žalúdka, hrubého a tenkého čreva a pečene.
Príznaky cystická fibróza
Cystická fibróza sa prejavuje rôznymi klinickými príznakmi. U novorodencov sa ochorenie môže prejaviť mekóniovou ileus. V dôsledku nedostatku alebo dokonca úplnej absencie trypsínu sa mekónium stáva veľmi hustým, viskóznym a hromadí sa v ileocekálnej oblasti. Ďalej sa vyvíja črevná obštrukcia, ktorá sa prejavuje intenzívnym vracaním s prímesou žlče, nadúvaním brucha, nedostatkom vylučovania mekónia, rozvojom príznakov peritonitídy a rýchlym zhoršením klinických prejavov syndrómu závažnej intoxikácie. Dieťa môže zomrieť v prvých dňoch života, ak sa nevykoná urgentný chirurgický zákrok.
V menej závažných prípadoch je charakteristickým znakom cystickej fibrózy bohatá, častá stolica, mastná, s veľkým množstvom tuku, s veľmi nepríjemným zápachom. U 1/3 pacientov sa pozoruje prolaps konečníka.
Následne sa u pacientov naďalej prejavuje črevná dysfunkcia, syndróm malabsorpcie, závažné poruchy fyzického vývoja a závažná hypovitaminóza.
V prvom alebo druhom roku života sa objavujú príznaky poškodenia bronchopulmonálneho systému (mierna forma ochorenia), ktoré sa prejavujú kašľom, ktorý môže byť extrémne výrazný a pripomínať kašeľ s čiernym kašľom. Kašeľ je sprevádzaný cyanózou, dýchavičnosťou a vylučovaním hustého spúta, spočiatku hlienového a potom hnisavého. Postupne sa vytvára klinický obraz chronickej obštrukčnej bronchitídy a bronchiektázie, pľúcneho emfyzému a respiračného zlyhania. Deti sú mimoriadne náchylné na akútne respiračné vírusové a bakteriálne infekcie, čo prispieva k exacerbáciám a progresii bronchopulmonálnej patológie. Možný je rozvoj bronchiálnej astmy závislej od infekcie.
U detí školského veku sa cystická fibróza môže prejaviť ako „črevná kolika“. Pacienti sa sťažujú na silnú paroxyzmálnu bolesť brucha, nadúvanie a opakované vracanie. Pri palpácii brucha sa určujú husté útvary, ktoré sa nachádzajú v projekcii hrubého čreva - stolica zmiešaná s hustým, hustým hlienom. Deti sú veľmi náchylné na rozvoj hypochloremickej alkalózy v dôsledku nadmerného vylučovania soli potom v horúcom počasí, pričom na pokožke dieťaťa sa objavuje „soľný mráz“.
Poruchy bronchopulmonálneho systému u dospelých
Poškodenie bronchopulmonálneho systému u pacientov s cystickou fibrózou (pľúcna forma ochorenia) je charakterizované rozvojom chronickej hnisavej obštrukčnej bronchitídy, bronchiektázie, chronickej pneumónie, pľúcneho emfyzému, respiračného zlyhania a pľúcneho srdcového ochorenia. U niektorých pacientov sa vyvinie pneumotorax a ďalšie komplikácie cystickej fibrózy: atelektáza, pľúcne abscesy, hemoptýza, pľúcne krvácanie a bronchiálna astma závislá od infekcie.
Pacienti sa sťažujú na bolestivý paroxyzmálny kašeľ s veľmi viskóznym, ťažko oddeliteľným hlienohnisavým spútom, niekedy s prímesou krvi. Okrem toho je mimoriadne charakteristická dýchavičnosť, najprv počas fyzickej námahy a potom v pokoji. Dýchavičnosť je spôsobená bronchiálnou obštrukciou. Mnoho pacientov sa sťažuje na chronickú nádchu spôsobenú polypózou a sinusitídou. Charakteristická je aj výrazná slabosť, progresívne znižovanie výkonu, časté akútne respiračné vírusové ochorenia. Pri vyšetrení sa pozornosť upriamuje na bledú pokožku, opuch tváre, cyanózu viditeľných slizníc a silnú dýchavičnosť. S rozvojom dekompenzovaného pľúcneho srdcového ochorenia sa objavujú opuchy v nohách. Môže sa pozorovať zhrubnutie koncových falangov prstov v tvare paličiek a nechtov v tvare hodinových skiel. Hrudník nadobúda tvar suda (v dôsledku rozvoja pľúcneho emfyzému).
Perkusia pľúc odhaľuje príznaky emfyzému - krabicový zvuk, prudké obmedzenie pohyblivosti pľúcneho okraja a zníženie dolného okraja pľúc. Auskultácia pľúc odhaľuje drsné dýchanie s predĺženým výdychom, rozptýlené suché sipoty a vlhké stredne silné a jemne bublajúce sipoty. Pri ťažkom emfyzéme pľúc je dýchanie prudko oslabené.
Extrapulmonálne prejavy cystickej fibrózy
Extrapulmonálne prejavy cystickej fibrózy môžu byť dosť výrazné a vyskytujú sa často.
Poškodenie pankreasu
U 85 % pacientov s cystickou fibrózou sa pozoruje insuficiencia exokrinnej funkcie pankreasu rôzneho stupňa závažnosti. Pri miernom poškodení pankreasu chýbajú syndrómy maldigescie a malabsorpcie, vyskytujú sa iba laboratórne prejavy exokrinnej insuficiencie (nízke hladiny trypsínu a lipázy v krvi a dvanástnikovom obsahu; často závažná steatorea). Je známe, že na prevenciu syndrómu maldigescie stačí sekrécia iba 1 až 2 % celkovej lipázy. Klinicky sa prejavujú iba významné poruchy exokrinnej funkcie.
Za normálnych podmienok produkujú acini pankreasu tekutý sekrét bohatý na enzýmy. Ako sa sekrét pohybuje pozdĺž vylučovacieho kanála, obohacuje sa o vodu a anióny a stáva sa ešte tekutejším. Pri cystickej fibróze, v dôsledku poruchy štruktúry a funkcie transmembránového regulátora (chloridového kanála), pankreatický sekrét nedostáva dostatočné množstvo tekutiny, stáva sa viskóznym a rýchlosť jeho pohybu pozdĺž vylučovacieho kanála sa prudko spomaľuje. Bielkoviny sekrétu sa ukladajú na stenách malých vylučovacích kanálov, čo vedie k ich obštrukcii. S postupom ochorenia sa nakoniec vyvíja deštrukcia a atrofia acini - vzniká chronická pankreatitída s exokrinnou pankreatickou insuficienciou. To sa klinicky prejavuje rozvojom syndrómov maldigescie a malabsorpcie. Pankreatická insuficiencia je hlavnou príčinou malabsorpcie tukov pri cystickej fibróze, ale zvyčajne sa pozoruje pri významnom deficite lipázy. Forsher a Durie (1991) uvádzajú, že pri úplnej absencii pankreatickej lipázy sa tuk rozkladá a vstrebáva z 50 – 60 %, čo je spôsobené prítomnosťou žalúdočných a slinných (sublingválnych) lipáz, ktorých aktivita sa blíži k dolnej hranici normy. Spolu s narušením rozkladu a vstrebávania tukov dochádza k narušeniu rozkladu a reabsorpcie bielkovín. Približne 50 % bielkovín prijatých s potravou sa stráca stolicou. Absorpcia sacharidov je napriek nedostatku α-amylázy ovplyvnená v menšej miere, ale metabolizmus sacharidov môže byť výrazne narušený.
Poškodenie pankreasu sa prejavuje vývojom syndrómu maldigescie a malabsorpcie s výrazným úbytkom hmotnosti a bohatou mastnou stolicou.
Vývoj syndrómov maldigescie a malabsorpcie je tiež uľahčený závažnou dysfunkciou črevných žliaz, zhoršenou sekréciou črevnej šťavy a znížením obsahu črevných enzýmov v nej.
Syndrómy maldigescie a malabsorpcie sa tiež nazývajú črevná forma cystickej fibrózy.
U pacientov s cystickou fibrózou v neskorších štádiách ochorenia (u 2 % detí a 15 % dospelých) sa pozoruje zhoršená endokrinná funkcia pankreasu (diabetes mellitus).
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Poškodenie pečene a žlčových ciest
U 13 % pacientov so zmiešanou a črevnou formou cystickej fibrózy sa vyvíja cirhóza pečene. Najtypickejšia je pre mutácie W128X, delta-P508 a X1303K. Biliárna cirhóza pečene s portálnou hypertenziou sa zisťuje u 5 – 10 % pacientov. Podľa Welcha, Smitha (1995) sa klinické, morfologické, laboratórne a inštrumentálne príznaky poškodenia pečene zisťujú u 86 % pacientov s cystickou fibrózou.
U mnohých pacientov s cystickou fibrózou sa tiež vyvinie chronická cholecystitída, často s kamennou formou.
Dysfunkcia pohlavných žliaz
U pacientov s cystickou fibrózou sa môže vyskytnúť azoospermia, ktorá je príčinou neplodnosti. Znížená plodnosť je typická aj pre ženy.
Etapy
Existujú tri stupne závažnosti pľúcnej cystickej fibrózy.
Mierna forma cystickej fibrózy sa vyznačuje zriedkavými exacerbáciami (nie viac ako raz ročne); počas období remisie klinické prejavy prakticky chýbajú a pacienti sú schopní pracovať.
Stredne závažné - exacerbácie sa pozorujú 2-3 krát ročne a trvajú približne 2 mesiace alebo dlhšie. Vo fáze exacerbácie sa vyskytuje intenzívny kašeľ s ťažko oddeľujúcim sa spútom, dýchavičnosť aj pri miernej fyzickej námahe, subfebrilná telesná teplota, celková slabosť, potenie. Zároveň dochádza k porušeniu exokrinnej funkcie pankreasu. Vo fáze remisie sa pracovná kapacita úplne neobnoví, dýchavičnosť počas fyzickej námahy pretrváva.
Závažný priebeh sa vyznačuje veľmi častými exacerbáciami ochorenia. Remisie prakticky chýbajú. V klinickom obraze sa do popredia dostávajú závažné respiračné zlyhanie, príznaky chronického pľúcneho srdcového ochorenia, často dekompenzovaného, typická je hemoptýza. Pozoruje sa výrazný úbytok hmotnosti, pacienti sú úplne invalidní. Závažnú bronchopulmonálnu patológiu spravidla sprevádza výrazná dysfunkcia pankreasu.
Formuláre
- Bronchopulmonálne lézie
- Opakovaná a recidivujúca pneumónia s predĺženým priebehom.
- Abscesujúca pneumónia, najmä u dojčiat.
- Chronická pneumónia, najmä bilaterálna.
- Bronchiálna astma refraktérna na tradičnú liečbu.
- Opakujúca sa bronchitída, bronchiolitída, najmä s kultúrou Pseudomonas aeruginosa.
- Zmeny v gastrointestinálnom trakte
- Mekóniový ileus a jeho ekvivalenty.
- Syndróm zhoršenej črevnej absorpcie neznámeho pôvodu.
- Obštrukčná žltačka u novorodencov s predĺženým priebehom.
- Cirhóza pečene.
- Diabetes mellitus.
- Gastroezofageálny reflux.
- Cholelitiáza.
- Rektálny prolaps.
- Zmeny v iných orgánoch a systémoch
- Poruchy rastu a vývoja.
- Oneskorený sexuálny vývoj.
- Mužská neplodnosť.
- Nosové polypy.
- Súrodenci z rodín s cystickou fibrózou.
[ 24 ]
Komplikácie a následky
Medzi komplikácie z gastrointestinálneho traktu patria:
- Diabetes mellitus sa vyvíja u 8 – 12 % pacientov starších ako 25 rokov.
- Fibrotizujúca kolonopatia.
- Mekóniový ileus v novorodeneckom období (u 12 % novorodencov s cystickou fibrózou), syndróm distálnej črevnej obštrukcie, rektálny prolaps, peptický vred a gastroezofageálna refluxná choroba.
Komplikácie pečene:
- Mastné ochorenie pečene (u 30 – 60 % pacientov),
- Fokálna biliárna cirhóza, multinodulárna biliárna cirhóza a pridružená portálna hypertenzia.
Portálna hypertenzia niekedy vedie k smrti v dôsledku pažerákových varixov.
Prevalencia cholecystitídy a žlčových kameňov je vyššia u pacientov s cystickou fibrózou ako u iných jedincov.
Oneskorená puberta a znížená plodnosť a ďalšie komplikácie. Väčšina mužov má azoospermiu a nedostatočný vývoj semenovodu.
Diagnostika cystická fibróza
Všeobecný krvný test - typická je anémia rôzneho stupňa závažnosti, zvyčajne normo- alebo hypochromická. Anémia má polyfaktoriálny pôvod (znížená absorpcia železa a vitamínu B12 v čreve v dôsledku rozvoja malabsorpčného syndrómu). Možná je leukopénia, s rozvojom hnisavej bronchitídy a pneumónie - leukocytóza, zvýšená sedimentácia erytrocytov.
Všeobecná analýza moču - žiadne významné zmeny, v zriedkavých prípadoch sa pozoruje mierna proteinúria.
Koprologické vyšetrenie - pozoruje sa steatorea, kreatorea. Becker (1987) odporúča meranie chymotrypsínu a mastných kyselín v stolici. Pred stanovením chymotrypsínu v stolici je potrebné prestať užívať tráviace enzýmy aspoň 3 dni pred vyšetrením. Pri cystickej fibróze je množstvo chymotrypsínu v stolici znížené a množstvo mastných kyselín zvýšené (normálne vylučovanie mastných kyselín je menej ako 20 mmol/deň). Je potrebné vziať do úvahy, že zvýšené vylučovanie mastných kyselín stolicou sa pozoruje aj u:
- nedostatok konjugovaných mastných kyselín v tenkom čreve v dôsledku zlyhania pečene, obštrukcie žlčovodov, významnej bakteriálnej kolonizácie tenkého čreva (v tomto prípade dochádza k intenzívnej hydrolýze žlčových kyselín);
- ileitída;
- celiakia (s rozvojom malabsorpčného syndrómu);
- enteritída;
- črevné lymfómy;
- Whippleova choroba;
- potravinové alergie;
- zrýchlený tranzit potravinových hmôt pri hnačke rôzneho pôvodu, karcinoidnom syndróme, tyreotoxikóze.
Biochemický krvný test - znížená hladina celkových bielkovín a albumínu, zvýšené hladiny alfa2 a gama globulínov, bilirubínu a aminotransferáz (v prípade poškodenia pečene), znížená aktivita amylázy, lipázy, trypsínu a hladiny železa a vápnika (v prípade vzniku syndrómu maldigescie, malabsorpcie).
Analýza spúta - prítomnosť veľkého počtu neutrofilných leukocytov a mikroorganizmov (počas bakterioskopie spúta).
Štúdia absorpčnej funkcie tenkého čreva a exokrinnej funkcie pankreasu odhaľuje významné poruchy.
Röntgenové vyšetrenie pľúc - odhaľuje zmeny, ktorých závažnosť závisí od závažnosti a fázy ochorenia. Najcharakteristickejšie zmeny sú:
- zvýšené pľúcne vzory v dôsledku peribronchiálnych intersticiálnych zmien;
- rozšírenie koreňov pľúc;
- obraz lobulárnej, subsegmentálnej alebo dokonca segmentálnej atelektázy pľúc;
- zvýšená priehľadnosť pľúcnych polí, najmä v horných častiach, nízka poloha a nedostatočná pohyblivosť bránice, rozšírenie retrosternálneho priestoru (prejav pľúcneho emfyzému);
- segmentálna alebo polysegmentálna infiltrácia pľúcneho tkaniva (pri rozvoji pneumónie).
Bronchografia odhaľuje zmeny spôsobené obštrukciou priedušiek viskóznym spútom (fragmentácia výplne priedušiek kontrastom, nerovnomerné kontúry, fenomén ruptúry priedušiek, výrazný pokles počtu laterálnych vetiev), ako aj bronchoekgázy (valcovité, zmiešané), lokalizované hlavne v dolných častiach pľúc.
Bronchoskopia odhaľuje difúznu hnisavú bronchitídu s bohatým hustým, viskóznym spútom a fibrínovými filmami.
Spirometria - už v počiatočných štádiách ochorenia odhaľuje respiračné zlyhanie obštrukčného typu (znížená FVC, FEV1, Tiffnov index), reštriktívne (znížená FVC) alebo najčastejšie obštrukčno-reštriktívne (znížená FVC, FVC, FEV1, Tiffnov index).
Gibsonov a Cookov potný test (test elektrolytov v pote) zahŕňa stimuláciu potenia pomocou elektroforézy s pilokarpínom s následným stanovením chloridov v pote. Doerehuk (1987) opisuje test nasledovne. Elektroforéza s pilokarpínom sa vykonáva na predlaktí, elektrický prúd je 3 mA. Po očistení pokožky destilovanou vodou sa pot zachytáva pomocou filtračného papiera umiestneného na stimulovanej oblasti, prikrytého gázou, aby sa zabránilo odparovaniu z nej. Po 30 – 60 minútach sa filtračný papier odstráni a eluuje sa v destilovanej vode. Meria sa množstvo zachyteného potu. Na dosiahnutie spoľahlivých výsledkov je potrebné zachytiť aspoň 50 mg (najlepšie 100 mg) potu.
Ak je koncentrácia chloridov vyššia ako 60 mmol/l, diagnóza cystickej fibrózy sa považuje za pravdepodobnú; ak je koncentrácia chloridov vyššia ako 100 mmol/l, je spoľahlivá; v tomto prípade by rozdiel v koncentrácii chlóru a sodíka nemal presiahnuť 8-10 mmol/l. Hadson (1983) odporúča, aby sa pri hraničnom obsahu sodíka a chloridov v pote vykonal prednizolónový test (5 mg perorálne počas 2 dní, po ktorom nasleduje stanovenie elektrolytov v pote). U jedincov, ktorí netrpia cystickou fibrózou, hladina sodíka v pote klesá na dolnú hranicu normálu; pri cystickej fibróze sa nemení. Potný test sa odporúča každému dieťaťu s chronickým kašľom.
Analýza krvných škvŕn alebo vzoriek DNA na prítomnosť hlavných mutácií génu cystickej fibrózy je najcitlivejším a najšpecifickejším diagnostickým testom. Táto metóda je však vhodná iba pre krajiny, kde je miera mutácií delta-P508 vyššia ako 80 %. Okrem toho je táto technika veľmi drahá a technicky zložitá.
Prenatálna diagnostika cystickej fibrózy sa vykonáva stanovením izoenzýmov alkalickej fosfatázy v plodovej vode. Táto metóda je možná od 18. do 20. týždňa tehotenstva.
Hlavné kritériá pre diagnostiku cystickej fibrózy sú nasledovné:
- indikácie v anamnéze oneskoreného fyzického vývoja v detstve, opakujúce sa chronické respiračné ochorenia, dyspeptické poruchy a hnačka, prítomnosť cystickej fibrózy u blízkych príbuzných;
- chronická obštrukčná bronchitída, často recidivujúca, s rozvojom bronchiektázie a pľúcneho emfyzému, často recidivujúca pneumónia;
- chronická recidivujúca pankreatitída s výrazným znížením exokrinnej funkcie, malabsorpčný syndróm;
- zvýšený obsah chlóru v pote pacienta;
- neplodnosť so zachovanou sexuálnou funkciou.
Úspešnú diagnostiku a diferenciálnu diagnostiku cystickej fibrózy uľahčuje identifikácia rizikových skupín.
Program skríningu cystickej fibrózy
- Všeobecná analýza krvi, moču, spúta.
- Bakteriologická analýza spúta.
- Koprologická analýza.
- Biochemický krvný test: stanovenie celkového proteínu a proteínových frakcií, glukózy, bilirubínu, aminotransferáz, alkalickej fosfatázy, gama-glutamyltranspeptidáz, draslíka, vápnika, železa, lipázy, amylázy, trypsínu.
- Štúdium exokrinnej funkcie pankreasu a absorpčnej funkcie čreva.
- Fluoroskopia a rádiografia pľúc, CT vyšetrenie pľúc.
- EKG.
- Echokardiografia.
- Bronchoskopia a bronchografia.
- Spirometria.
- Potný test.
- Konzultácia s genetikom.
- Analýza krvných škvŕn alebo vzoriek DNA na prítomnosť hlavných mutácií génu cystickej fibrózy.
Aké testy sú potrebné?
Komu sa chcete obrátiť?
Liečba cystická fibróza
Typ a závažnosť príznakov cystickej fibrózy sa môžu značne líšiť, takže neexistuje typický liečebný plán; je individualizovaný pre každého jednotlivca.
Terapia pozostáva z nasledujúcich terapeutických opatrení:
- Dýchacie cvičenia a posturálna drenáž pomáhajú zbaviť sa hustého hlienu, ktorý sa hromadí v pľúcach. Niektoré techniky uvoľňovania dýchacích ciest vyžadujú pomoc od členov rodiny, priateľov alebo pneumológa. Mnoho ľudí používa nafukovaciu hrudnú vestu, ktorá vibruje na vysokej frekvencii.

- Inhalačné lieky, ktoré majú bronchodilatačné, drenážne (mukolytiká) a antibakteriálne účinky (napríklad fluorochinolóny).
- Prípravky obsahujúce pankreatické enzýmy na zlepšenie trávenia. Tieto prípravky sa užívajú počas jedla.
- Multivitamíny (vrátane vitamínov rozpustných v tukoch).
V roku 2015 schválil Úrad pre kontrolu potravín a liečiv (FDA) druhý liek na liečbu cystickej fibrózy, ktorý je zameraný na defektný proteín známy ako CFTR. Prvý liek, takzvaný modulátor CFTR, bol schválený v roku 2012. Očakáva sa, že modulátory CFTR predĺžia životy niektorých ľudí s cystickou fibrózou o desaťročia.
Na liečbu nasledujúcich respiračných komplikácií môže byť potrebný chirurgický zákrok:
- Pneumotorax, masívna recidivujúca alebo pretrvávajúca hemoptýza, nosové polypy, pretrvávajúca a chronická sinusitída.
- Mekóniový ileus, invaginácia čreva, rektálny prolaps.
Transplantácia pľúc sa vykonáva v terminálnom štádiu ochorenia.
Predpoveď
Priemerný vek prežitia pacientov s cystickou fibrózou sa pohybuje od 35 do 40 rokov. Priemerný vek prežitia je vyšší u mužov ako u žien.
Vďaka moderným liečebným stratégiám sa 80 % pacientov dožije dospelosti. Cystická fibróza však výrazne obmedzuje funkčné schopnosti pacienta. Na toto ochorenie stále neexistuje liek.