Lekársky expert článku

Nové publikácie
Dedičná nefritída (Alportov syndróm) u detí
Posledná kontrola: 05.07.2025

Všetok obsah iLive je lekársky kontrolovaný alebo kontrolovaný, aby sa zabezpečila čo najväčšia presnosť faktov.
Máme prísne smernice týkajúce sa získavania zdrojov a len odkaz na seriózne mediálne stránky, akademické výskumné inštitúcie a vždy, keď je to možné, na lekársky partnerské štúdie. Všimnite si, že čísla v zátvorkách ([1], [2] atď.) Sú odkazmi na kliknutia na tieto štúdie.
Ak máte pocit, že niektorý z našich obsahov je nepresný, neaktuálny alebo inak sporný, vyberte ho a stlačte kláves Ctrl + Enter.
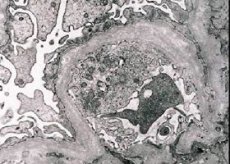
Hereditárna nefritída (Alportov syndróm) je geneticky podmienená dedičná neimunitná glomerulopatia, ktorá sa prejavuje hematúriou (niekedy s proteinúriou), progresívnym poklesom funkcie obličiek s rozvojom chronického zlyhania obličiek, často kombinovaným so senzorineurálnou hluchotou a zrakovým postihnutím.
Ochorenie prvýkrát opísal v roku 1902 LG Guthrie, ktorý pozoroval rodinu, v ktorej sa hematúria pozorovala v niekoľkých generáciách. V roku 1915 A. F. Hurst opísal vývoj urémie u členov tej istej rodiny. V roku 1927 A. Alport prvýkrát identifikoval stratu sluchu u niekoľkých príbuzných s hematúriou. V 50. rokoch 20. storočia boli opísané očné lézie pri podobnom ochorení. V roku 1972 u pacientov s dedičnou hematúriou počas morfologickej štúdie renálneho tkaniva Hinglais a kol. odhalili nerovnomerné rozšírenie a stratifikáciu glomerulárnych bazálnych membrán. V roku 1985 bol identifikovaný genetický základ dedičnej nefritídy - mutácia v géne kolagénu typu IV (Fiengold a kol., 1985).
Štúdium genetickej povahy ochorenia nám umožnilo dospieť k záveru, že rozdiely vo fenotypových prejavoch dedičnej nefritídy (s poruchou sluchu alebo bez nej) sú spôsobené stupňom expresie mutantného génu. V súčasnosti sa teda všetky klinické varianty považujú za prejavy jednej choroby a termín „dedičná nefritída“ je synonymom termínu „Alportov syndróm“.
Podľa epidemiologických štúdií sa dedičná nefritída vyskytuje s frekvenciou 17 na 100 000 detí.
 [
[ Príčiny Alportovho syndrómu
Genetickým základom ochorenia je mutácia v géne reťazca a-5 kolagénu typu IV. Tento typ je univerzálny pre bazálne membrány obličiek, kochleárny aparát, puzdro šošovky, sietnicu a rohovku oka, čo bolo dokázané v štúdiách s použitím monoklonálnych protilátok proti tejto frakcii kolagénu. Nedávno sa ukázala možnosť použitia DNA sond na prenatálnu diagnostiku dedičnej nefritídy.
Zdôrazňuje sa dôležitosť testovania všetkých členov rodiny pomocou DNA sond na identifikáciu nositeľov mutantného génu, čo má veľký význam pri vykonávaní lekárskeho a genetického poradenstva rodín s týmto ochorením. Až 20 % rodín však nemá príbuzných trpiacich ochorením obličiek, čo naznačuje vysokú frekvenciu spontánnych mutácií abnormálneho génu. Väčšina pacientov s dedičnou nefritídou má vo svojich rodinách osoby s ochorením obličiek, stratou sluchu a poruchami zraku; dôležité sú pokrvné manželstvá medzi ľuďmi s jedným alebo viacerými predkami, pretože v manželstve príbuzných osôb sa zvyšuje pravdepodobnosť prijatia rovnakých génov od oboch rodičov. Boli stanovené autozomálne dominantné, autozomálne recesívne a dominantné, X-viazané cesty prenosu.
U detí sa najčastejšie rozlišujú tri typy dedičnej nefritídy: Alportov syndróm, dedičná nefritída bez straty sluchu a familiárna benígna hematúria.
Alportov syndróm je dedičná nefritída so sluchovým postihnutím. Jej základom je kombinovaná porucha štruktúry kolagénu glomerulárnej bazálnej membrány obličiek, štruktúr ucha a oka. Gén klasického Alportovho syndrómu sa nachádza v lokuse 21-22 q dlhého ramena chromozómu X. Vo väčšine prípadov sa dedí dominantným spôsobom, viazaný na chromozóm X. V tomto ohľade je Alportov syndróm závažnejší u mužov, pretože u žien je funkcia mutantného génu kompenzovaná zdravou alelou druhého, nepoškodeného chromozómu.
Genetickým základom pre vznik dedičnej nefritídy sú mutácie v génoch alfa reťazcov kolagénu typu IV. Je známych šesť alfa reťazcov kolagénu typu IV G: gény reťazcov a5 a a6 (Col4A5 a Col4A5) sa nachádzajú na dlhom ramene chromozómu X v zóne 21-22q; gény reťazcov a3 a a4 (Col4A3 a Col4A4) sú na 2. chromozóme; gény reťazcov a1 a a2 (Col4A1 a Col4A2) sú na 13. chromozóme.
Vo väčšine prípadov (80 – 85 %) sa zisťuje X-viazaný typ dedičnosti ochorenia, spojený s poškodením génu Col4A5 v dôsledku delécie, bodových mutácií alebo porúch zostrihu. V súčasnosti bolo nájdených viac ako 200 mutácií génu Col4A5, ktoré sú zodpovedné za narušenie syntézy a5-reťazcov kolagénu typu IV. Pri tomto type dedičnosti sa ochorenie prejavuje u detí oboch pohlaví, ale u chlapcov je závažnejšie.
Mutácie v lokusoch génov Col4A3 a Col4A4 zodpovedných za syntézu reťazcov a3 a a4 kolagénu typu IV sa dedia autozomálne. Podľa výskumu sa autozomálne dominantný typ dedičnosti pozoruje u 16 % prípadov dedičnej nefritídy a autozomálne recesívny typ sa pozoruje u 6 % pacientov. Je známych približne 10 variantov mutácií génov Col4A3 a Col4A4.
Výsledkom mutácií je porušenie procesov montáže kolagénu typu IV, čo vedie k narušeniu jeho štruktúry. Kolagén typu IV je jednou z hlavných zložiek glomerulárnej bazálnej membrány, kochleárneho aparátu a očnej šošovky, ktorej patológia bude zistená v klinike dedičnej nefritídy.
Kolagén typu IV, ktorý je súčasťou glomerulárnej bazálnej membrány, pozostáva prevažne z dvoch reťazcov a1 (IV) a jedného reťazca a2 (IV) a obsahuje aj reťazce a3, a4, a5. Najčastejšie je pri X-viazanej dedičnosti mutácia génu Col4A5 sprevádzaná absenciou reťazcov a3, a4, a5 a a6 v štruktúre kolagénu typu IV a počet reťazcov o1 a a2 v glomerulárnej bazálnej membráne sa zvyšuje. Mechanizmus tohto javu nie je jasný, predpokladá sa, že príčinou sú posttranskripčné zmeny v mRNA.
Absencia reťazcov a3, a4 a a5 v štruktúre kolagénu typu IV glomerulárnych bazálnych membrán vedie k ich stenčeniu a krehkosti v skorých štádiách Alportovho syndrómu, čo sa klinicky prejavuje častejšie hematúriou (menej často hematúriou s proteinúriou alebo iba proteinúriou), stratou sluchu a lentikonom. Ďalšia progresia ochorenia vedie v neskorších štádiách ochorenia k zhrubnutiu a zhoršenej permeabilite bazálnych membrán s proliferáciou kolagénu typu V a VI v nich, čo sa prejavuje zvýšením proteinúrie a znížením funkcie obličiek.
Povaha mutácie, ktorá je základom dedičnej nefritídy, do značnej miery určuje jej fenotypový prejav. V prípade delécie chromozómu X so súčasnou mutáciou génov Col4A5 a Col4A6 zodpovedných za syntézu a5- a a6-reťazcov kolagénu typu IV sa Alportov syndróm kombinuje s leiomyomatózou pažeráka a genitálií. Podľa výskumných údajov sa v prípade mutácie génu Col4A5 spojenej s deléciou zaznamenáva väčšia závažnosť patologického procesu, kombinácia poškodenia obličiek s extrarenálnymi prejavmi a skorým rozvojom chronického zlyhania obličiek v porovnaní s bodovou mutáciou tohto génu.
Morfologicky elektrónová mikroskopia odhaľuje stenčenie a stratifikáciu glomerulárnych bazálnych membrán (najmä lamina densa) a prítomnosť elektrónovo denzných granúl. Glomerulárne lézie môžu byť u toho istého pacienta heterogénne, od minimálnych fokálnych mezangiálnych lézií až po glomerulosklerózu. Glomerulitída pri Alportovom syndróme je vždy imunonegatívna, čo ju odlišuje od glomerulonefritídy. Medzi charakteristické znaky patrí rozvoj tubulárnej atrofie, lymfohistiocytová infiltrácia a prítomnosť „penových buniek“ s lipidovými inklúziami – lipofágov. S progresiou ochorenia sa odhaľuje zhrubnutie a výrazná deštrukcia glomerulárnych bazálnych membrán.
Zistili sa určité zmeny v imunitnom systéme. Pacienti s dedičnou nefritídou majú zníženú hladinu Ig A a tendenciu k zvýšenej koncentrácii IgM v krvi, pričom hladina IgG môže byť zvýšená v skorých štádiách ochorenia a znížená v neskorších štádiách. Možno je zvýšenie koncentrácie IgM a G druhom kompenzačnej reakcie v reakcii na nedostatok IgA.
Funkčná aktivita T-lymfocytového systému je znížená; zaznamenáva sa selektívny pokles B-lymfocytov zodpovedných za syntézu Ig A, fagocytárna väzba imunity je narušená, najmä v dôsledku narušenia chemotaxy a intracelulárnych tráviacich procesov v neutrofiloch.
Pri vyšetrení biopsie obličiek u pacientov s Alportovým syndrómom údaje z elektrónovej mikroskopie odhaľujú ultraštrukturálne zmeny v glomerulárnej bazálnej membráne: stenčenie, narušenie štruktúry a štiepenie glomerulárnych bazálnych membrán so zmenou ich hrúbky a nerovnými kontúrami. V skorých štádiách dedičnej nefritídy defekt určuje stenčenie a krehkosť glomerulárnych bazálnych membrán.
Riedenie glomerulárnych membrán je priaznivejším znakom a je častejšie u dievčat. Konštantnejším elektrónmikroskopickým znakom pri dedičnej nefritíde je štiepenie bazálnej membrány a závažnosť jej deštrukcie koreluje so závažnosťou procesu.
Príznaky Alportovho syndrómu u detí
Prvé príznaky Alportovho syndrómu vo forme izolovaného močového syndrómu sa najčastejšie zisťujú u detí v prvých troch rokoch života. Vo väčšine prípadov sa ochorenie zistí náhodne. Močový syndróm sa zistí počas preventívnej prehliadky dieťaťa, pred prijatím do zariadenia starostlivosti o deti alebo počas ARVI. V prípade patológie v moči počas ARVI. Pri dedičnej nefritíde, na rozdiel od získanej glomerulonefritídy, neexistuje latentné obdobie.
V počiatočnom štádiu ochorenia je zdravie dieťaťa málo ohrozené, charakteristickým znakom je pretrvávanie a odolnosť močového syndrómu. Jedným z hlavných príznakov je hematúria rôzneho stupňa závažnosti, pozorovaná v 100 % prípadov. Zvýšenie stupňa hematúrie sa zaznamenáva počas alebo po respiračných infekciách, fyzickej aktivite alebo po preventívnych očkovaniach. Proteinúria vo väčšine prípadov nepresahuje 1 g/deň, na začiatku ochorenia môže byť nekonštantná, s postupom procesu sa proteinúria zvyšuje. Periodicky môže byť v močovom sedimente prítomná leukocytúria s prevahou lymfocytov, čo je spojené s rozvojom intersticiálnych zmien.
Následne dochádza k čiastočnej poruche funkcie obličiek, zhoršuje sa celkový stav pacienta: objavuje sa intoxikácia, svalová slabosť, arteriálna hypotenzia, často sa objavuje porucha sluchu (najmä u chlapcov) a niekedy aj porucha zraku. Intoxikácia sa prejavuje bledosťou, únavou a bolesťami hlavy. V počiatočnom štádiu ochorenia sa strata sluchu vo väčšine prípadov zistí iba audiografiou. Strata sluchu pri Alportovom syndróme sa môže vyskytnúť v rôznych obdobiach detstva, ale najčastejšie sa strata sluchu diagnostikuje vo veku 6 – 10 rokov. Strata sluchu u detí začína vysokými frekvenciami, dosahuje významný stupeň vo vzdušnom a kostnom vedení, prechádza zo straty sluchu na vedenie zvuku do straty sluchu vnímajúcej zvuk. Strata sluchu môže byť jedným z prvých príznakov ochorenia a môže predchádzať močovému syndrómu.
V 20 % prípadov majú pacienti s Alportovým syndrómom zmeny v zrakových orgánoch. Najčastejšie zistenými anomáliami sú anomálie šošovky: sferofókia, predný, zadný alebo zmiešaný lentikonus a rôzne katarakty. V rodinách s Alportovým syndrómom je značný výskyt krátkozrakosti. Mnohí výskumníci v týchto rodinách neustále zaznamenávajú bilaterálne perimakulárne zmeny vo forme jasných belavých alebo žltkastých granulácií v žltom telese. Tento príznak považujú za konštantný symptóm, ktorý má pri Alportovom syndróme vysokú diagnostickú hodnotu. KS Chugh a kol. (1993) v oftalmologickej štúdii zistili u pacientov s Alportovým syndrómom zníženie zrakovej ostrosti v 66,7 % prípadov, predný lentikonus v 37,8 %, retinálne škvrny v 22,2 %, kataraktu v 20 % a keratokonus v 6,7 %.
U niektorých detí s dedičnou nefritídou, najmä pri vzniku zlyhania obličiek, sa pozoruje výrazné oneskorenie vo fyzickom vývoji. S progresiou zlyhania obličiek sa vyvíja arteriálna hypertenzia. U detí sa častejšie zisťuje v dospievaní a vo vyšších vekových skupinách.
Pacienti s dedičnou nefritídou sa vyznačujú prítomnosťou rôznych (viac ako 5-7) stigmi dysmorfogenézy spojivového tkaniva. Medzi stigmami spojivového tkaniva u pacientov sú najčastejšie hypertelorizmus očí, vysoké podnebie, anomálie zhryzu, abnormálny tvar ušných boltcov, zakrivenie malíčka na rukách a „sandálová medzera“ na nohách. Dedičná nefritída sa vyznačuje jednotnosťou stigmi dysmorfogenézy v rámci rodiny, ako aj vysokou frekvenciou ich rozšírenia medzi príbuznými probandov, po ktorých línii sa ochorenie prenáša.
V skorých štádiách ochorenia sa zisťuje izolované zníženie parciálnych renálnych funkcií: transport aminokyselín, elektrolytov, koncentračná funkcia, acidogenéza, neskoršie zmeny ovplyvňujú funkčný stav proximálnej aj distálnej časti nefrónu a sú charakterizované kombinovanými parciálnymi poruchami. Pokles glomerulárnej filtrácie nastáva neskôr, častejšie v adolescencii. S progresiou dedičnej nefritídy sa vyvíja anémia.
Hereditárna nefritída sa teda vyznačuje postupným priebehom ochorenia: najprv latentné štádium alebo skryté klinické príznaky, ktoré sa prejavujú minimálnymi zmenami močového syndrómu, potom dochádza k postupnej dekompenzácii procesu so znížením funkcie obličiek s manifestnými klinickými príznakmi (intoxikácia, asténia, vývojové oneskorenie, anémia). Klinické príznaky sa zvyčajne objavujú bez ohľadu na vrstvenie zápalovej reakcie.
Dedičná nefritída sa môže prejaviť v rôznych vekových obdobiach, čo závisí od pôsobenia génu, ktorý je do určitého času v potlačenom stave.
Klasifikácia
Existujú tri typy dedičnej nefritídy
- Možnosť I - klinicky sa prejavuje ako nefritída s hematúriou, stratou sluchu a poškodením očí. Priebeh nefritídy je progresívny s rozvojom chronického zlyhania obličiek. Typ dedičnosti je dominantný, viazaný na chromozóm X. Morfologicky sa odhaľuje porušenie štruktúry bazálnej membrány, jej stenčenie a štiepenie.
- Možnosť II - klinicky sa prejavuje ako nefritída s hematúriou bez straty sluchu. Priebeh nefritídy je progresívny s rozvojom chronického zlyhania obličiek. Typ dedičnosti je dominantný, viazaný na chromozóm X. Morfologicky sa zisťuje stenčenie glomerulárnej kapilárnej bazálnej membrány (najmä laminadensa).
- Možnosť III - benígna familiárna hematúria. Priebeh je priaznivý, chronické zlyhanie obličiek sa nevyvíja. Typ dedičnosti je autozomálne dominantný alebo autozomálne recesívny. Pri autozomálne recesívnom type dedičnosti sa u žien pozoruje závažnejší priebeh ochorenia.
Diagnóza Alportovho syndrómu
Navrhujú sa tieto kritériá:
- prítomnosť aspoň dvoch pacientov s nefropatiou v každej rodine;
- hematúria ako hlavný príznak nefropatie u probanta;
- prítomnosť straty sluchu u aspoň jedného člena rodiny;
- rozvoj chronického zlyhania obličiek u jedného alebo viacerých príbuzných.
V diagnostike rôznych dedičných a vrodených ochorení sa veľký priestor venuje komplexnému prístupu k vyšetreniu a predovšetkým venovaniu pozornosti údajom získaným pri zostavovaní rodokmeňa dieťaťa. Diagnóza Alportovho syndrómu sa považuje za platnú v prípadoch, keď sa u pacienta zistia 3 zo 4 typických znakov: prítomnosť hematúrie a chronického zlyhania obličiek v rodine, prítomnosť neurosenzorickej straty sluchu, patológia zraku u pacienta, detekcia znakov štiepenia glomerulárnej bazálnej membrány so zmenou jej hrúbky a nerovnými kontúrami počas elektrónmikroskopických charakteristík biopsie.
Vyšetrenie pacienta by malo zahŕňať klinické a genetické výskumné metódy; cielené štúdium anamnézy ochorenia; všeobecné vyšetrenie pacienta s prihliadnutím na diagnosticky významné kritériá. V štádiu kompenzácie možno patológiu zistiť iba zameraním sa na také syndrómy, ako je prítomnosť dedičnej záťaže, hypotenzia, viacnásobné stigmy dysembryogenézy, zmeny v močovom syndróme. V štádiu dekompenzácie sa môžu objaviť extrarenálne príznaky, ako je ťažká intoxikácia, asténia, oneskorený fyzický vývoj, anémia, ktoré sa prejavujú a zhoršujú s postupným znižovaním funkcie obličiek. U väčšiny pacientov so zníženou funkciou obličiek sa pozoruje: znížená acido- a aminogenéza; 50 % pacientov zaznamenáva významný pokles sekrečnej funkcie obličiek; obmedzený rozsah kolísania optickej hustoty moču; porucha filtračného rytmu a následne pokles glomerulárnej filtrácie. Štádium chronického zlyhania obličiek sa diagnostikuje, keď majú pacienti zvýšenú hladinu močoviny v krvnom sére (viac ako 0,35 g/l) počas 3-6 mesiacov alebo dlhšie a pokles glomerulárnej filtrácie na 25 % normy.
Diferenciálna diagnostika dedičnej nefritídy by sa mala vykonávať predovšetkým s hematurickou formou získanej glomerulonefritídy. Získaná glomerulonefritída má najčastejšie akútny nástup, obdobie 2-3 týždňov po infekcii, extrarenálne príznaky vrátane hypertenzie od prvých dní (pri dedičnej nefritíde naopak hypotenzia), zníženú glomerulárnu filtráciu na začiatku ochorenia, žiadne poškodenie čiastočných tubulárnych funkcií, zatiaľ čo pri dedičnej sú prítomné. Získaná glomerulonefritída sa prejavuje výraznejšou hematúriou a proteinúriou so zvýšenou sedimentáciou erytrocytov (ESR). Diagnostickú hodnotu majú typické zmeny v glomerulárnej bazálnej membráne, charakteristické pre dedičnú nefritídu.
Diferenciálna diagnostika od dysmetabolickej nefropatie sa vykonáva s chronickým zlyhaním obličiek, v rodine sa klinicky prejavujú heterogénne ochorenia obličiek a môže sa vyskytnúť spektrum nefropatie od pyelonefritídy až po urolitiázu. Deti sa často sťažujú na bolesť brucha a periodicky počas močenia, v močovom sedimente - oxaláty.
Ak existuje podozrenie na dedičnú nefritídu, pacient by mal byť odoslaný na špecializované nefrologické oddelenie, aby sa objasnila diagnóza.
Čo je potrebné preskúmať?
Ako preskúmať?
Aké testy sú potrebné?
Komu sa chcete obrátiť?
Liečba Alportovho syndrómu
Režim zahŕňa obmedzenia ťažkej fyzickej námahy a pobytu na čerstvom vzduchu. Strava je kompletná, s dostatočným množstvom kompletných bielkovín, tukov a sacharidov, pričom sa zohľadňuje funkcia obličiek. Veľký význam má detekcia a liečba chronických ložísk infekcie. Používajú sa nasledujúce lieky: ATP, kokarboxyláza, pyridoxín (do 50 mg/deň), karnitínchlorid. Kúry sa podávajú 2-3 krát ročne. Pri hematúrii sa predpisuje bylinná medicína - žihľava dvojdomá, šťava z arónie, rebríček.
V zahraničnej aj domácej literatúre existujú správy o liečbe prednizolónom a použití cytostatík. Je však ťažké posúdiť účinok.
Pri chronickom zlyhaní obličiek sa používa hemodialýza a transplantácia obličiek.
Neexistujú žiadne metódy špecifickej (účinnej patogenetickej) terapie dedičnej nefritídy. Všetky liečebné opatrenia sú zamerané na prevenciu a spomalenie poklesu funkcie obličiek.
Strava by mala byť vyvážená a kalorická, berúc do úvahy funkčný stav obličiek. Pri absencii funkčných porúch by detská strava mala obsahovať dostatok bielkovín, tukov a sacharidov. Pri výskyte príznakov renálnej dysfunkcie by sa malo obmedziť množstvo bielkovín, sacharidov, vápnika a fosforu, čo odďaľuje rozvoj chronického zlyhania obličiek.
Fyzická aktivita by mala byť obmedzená; deťom sa odporúča vyhýbať sa športu.
Treba sa vyhýbať kontaktu s infekčnými pacientmi, znížiť riziko vzniku akútnych respiračných ochorení. Je potrebná sanitácia ložísk chronickej infekcie. Preventívne očkovania sa u detí s dedičnou nefritídou nevykonávajú, očkovanie je možné len z epidemiologických dôvodov.
Hormonálna a imunosupresívna liečba pri dedičnej nefritíde je neúčinná. Existujú náznaky určitého pozitívneho účinku (zníženie proteinúrie a spomalenie progresie ochorenia) pri dlhodobom viacročnom užívaní cyklosporínu A a ACE inhibítorov.
Pri liečbe pacientov sa používajú lieky, ktoré zlepšujú metabolizmus:
- pyridoxín - 2-3 mg/kg/deň v 3 dávkach počas 4 týždňov;
- kokarboxyláza - 50 mg intramuskulárne každý druhý deň, celkovo 10-15 injekcií;
- ATP - 1 ml intramuskulárne každý druhý deň, 10-15 injekcií;
- vitamín A - 1000 IU/rok/deň v 1 dávke počas 2 týždňov;
- Vitamín E - 1 mg/kg/deň v 1 dávke počas 2 týždňov.
Tento typ terapie pomáha zlepšiť celkový stav pacientov, znížiť tubulárne dysfunkcie a vykonáva sa v kurzoch 3-krát ročne.
Levamizol sa môže použiť ako imunomodulátor - 2 mg/kg/deň 2-3 krát týždenne s prestávkami medzi dávkami 3-4 dni.
Podľa výskumných údajov má hyperbarická oxygenácia pozitívny vplyv na závažnosť hematúrie a renálnej dysfunkcie.
Najúčinnejšou metódou liečby dedičnej nefritídy je včasná transplantácia obličiek. V tomto prípade nedochádza k relapsu ochorenia v transplantovanej obličke; v malom percente prípadov (približne 5 %) sa v transplantovanej obličke môže vyvinúť nefritída spojená s antigénmi proti glomerulárnej bazálnej membráne.
Sľubným smerom je prenatálna diagnostika a genetické inžinierstvo. Experimenty na zvieratách ukazujú vysokú účinnosť prenosu normálnych génov zodpovedných za syntézu alfa reťazcov kolagénu typu IV do renálneho tkaniva, po čom sa pozoruje syntéza normálnych kolagénových štruktúr.
Predpoveď
Prognóza dedičnej nefritídy je vždy vážna.
Prognosticky nepriaznivé kritériá pre priebeh dedičnej nefritídy sú:
- mužské pohlavie;
- skorý rozvoj chronického zlyhania obličiek u rodinných príslušníkov;
- proteinúria (viac ako 1 g/deň);
- zhrubnutie glomerulárnych bazálnych membrán podľa mikroskopie;
- akustická neuritída;
- delécia v géne Col4A5.
Prognóza benígnej familiárnej hematúrie je priaznivejšia.
Использованная литература