Lekársky expert článku

Nové publikácie
Prióny - pôvodcovia priónových ochorení
Posledná kontrola: 06.07.2025

Všetok obsah iLive je lekársky kontrolovaný alebo kontrolovaný, aby sa zabezpečila čo najväčšia presnosť faktov.
Máme prísne smernice týkajúce sa získavania zdrojov a len odkaz na seriózne mediálne stránky, akademické výskumné inštitúcie a vždy, keď je to možné, na lekársky partnerské štúdie. Všimnite si, že čísla v zátvorkách ([1], [2] atď.) Sú odkazmi na kliknutia na tieto štúdie.
Ak máte pocit, že niektorý z našich obsahov je nepresný, neaktuálny alebo inak sporný, vyberte ho a stlačte kláves Ctrl + Enter.
Pomalé vírusové infekcie sa vyznačujú špeciálnymi kritériami:
- nezvyčajne dlhá inkubačná doba (mesiace, roky);
- špecifická lézia orgánov a tkanív, predovšetkým centrálneho nervového systému;
- pomalý, stabilný priebeh ochorenia;
- nevyhnutný smrteľný výsledok.

Niektoré patogény, ktoré spôsobujú akútne vírusové infekcie, môžu tiež spôsobovať pomalé vírusové infekcie. Napríklad vírus osýpok niekedy spôsobuje SSPE a vírus rubeoly spôsobuje progresívnu vrodenú rubeolu a rubeolovú panencefalitídu.
Typickú pomalú vírusovú infekciu u zvierat spôsobuje vírus visna/madi, čo je retrovírus. Je pôvodcom pomalej vírusovej infekcie a progresívnej pneumónie u oviec. Biela hmota mozgu je zničená, vyvíja sa paralýza (visna - chradnutie); dochádza k chronickému zápalu pľúc a sleziny.
Choroby, ktoré sa svojimi znakmi podobajú pomalým vírusovým infekciám, sú spôsobené priónmi - pôvodcami priónových infekcií. Priónové choroby sú skupinou progresívnych porúch centrálneho nervového systému ľudí a zvierat. U ľudí je funkcia centrálneho nervového systému narušená, dochádza k zmenám osobnosti a poruchám pohybu. Príznaky ochorenia zvyčajne trvajú niekoľko mesiacov až niekoľko rokov a končia smrťou. Predtým sa priónové infekcie považovali za spoločné s tzv. pôvodcami pomalých vírusových infekcií.
Niektoré agensy, ktoré spôsobujú priónové ochorenia, sa hromadia najskôr v lymfoidných tkanivách. Prióny sa po vstupe do mozgu hromadia vo veľkých množstvách a spôsobujú amyloidózu (extracelulárnu dysproteinózu, charakterizovanú ukladaním amyloidu s rozvojom atrofie a sklerózy tkaniva) a astrocytózu (proliferácia astrocytových neuroglií, hyperprodukcia gliových vlákien). Vznikajú fibrily, zhluky bielkovín alebo amyloidu a spongiformné zmeny v mozgu (prenosné spongiformné encefalopatie). V dôsledku toho sa mení správanie, zhoršuje sa koordinácia pohybov, vyvíja sa vyčerpanie s fatálnym koncom. Imunita sa netvorí. Priónové ochorenia sú konformačné ochorenia, ktoré sa vyvíjajú v dôsledku nesprávneho skladania (porušenia správnej konformácie) bunkového proteínu potrebného pre normálne fungovanie tela. Spôsoby prenosu priónov sú rôzne:
- alimentárna cesta - infikované produkty živočíšneho pôvodu, potravinárske prísady zo surových hovädzích orgánov atď.:
- prenos prostredníctvom transfúzie krvi, podávania liekov živočíšneho pôvodu, transplantácie orgánov a tkanív, použitia infikovaných chirurgických a zubných nástrojov;
- prenos prostredníctvom imunobiologických prípravkov (známa je infekcia 1500 oviec PrP''' vakcínou z formonoplastu mozgu z chorých oviec).
Patologické prióny sa po vstupe do čreva transportujú do krvi a lymfy. Po periférnej replikácii v slezine, slepom čreve, mandliach a iných lymfoidných tkanivách sa prenášajú do mozgu cez periférne nervy (neuroinvázia). Priamy prienik priónov do mozgu cez hematoencefalickú bariéru je možný. Predtým sa predpokladalo, že centrálny nervový systém je jediné tkanivo, v ktorom sa patologické prióny hromadia, ale objavili sa štúdie, ktoré túto hypotézu zmenili. Ukázalo sa, že hromadenie priónov v slezine je spojené so zvýšením počtu a fungovaním folikulárnych dendritických buniek.
 [
[ Vlastnosti priónov
Normálna bunková izoforma priónového proteínu s molekulovou hmotnosťou 33-35 kDa je určená génom priónového proteínu (priónový gén - PrNP sa nachádza na 20. ľudskom chromozóme). Normálny gén sa nachádza na povrchu bunky (ukotvený v membráne glykoproteínom molekuly), citlivý na proteázu. Reguluje prenos nervových impulzov, denné cykly, oxidačné procesy, podieľa sa na metabolizme medi v centrálnom nervovom systéme a na regulácii delenia kmeňových buniek kostnej drene. Okrem toho sa priónový gén nachádza v slezine, lymfatických uzlinách, koži, gastrointestinálnom trakte a folikulárnych dendritických bunkách.
Proliferácia patologických priónov
Transformácia priónov do zmenených foriem nastáva, keď je narušená kineticky riadená rovnováha medzi nimi. Proces je umocnený zvýšením množstva patologického (PrP) alebo exogénneho priónu. PrP je normálny proteín ukotvený v bunkovej membráne. PrP' je globulárny hydrofóbny proteín, ktorý tvorí agregáty sám so sebou a s PrP'' na povrchu bunky: v dôsledku toho sa PrP' transformuje na PrP'' a potom cyklus pokračuje. Patologická forma PrP''' sa hromadí v neurónoch, čo dáva bunke špongiovitý vzhľad.
Kuru
Priónová choroba, predtým bežná medzi Papuáncami (čo znamená trasenie alebo chvenie) vo východnej časti ostrova Nová Guinea. Infekčné vlastnosti choroby dokázal K. Gajdusek. Patogén sa prenáša potravou v dôsledku rituálneho kanibalizmu - konzumácie nedostatočne tepelne upraveného, priónmi infikovaného mozgu mŕtvych príbuzných. V dôsledku poškodenia centrálneho nervového systému dochádza k zhoršeniu pohybu a chôdze, objavuje sa zimnica a eufória („smejúca sa smrť“). Inkubačná doba trvá 5-30 rokov. Pacient zomiera po roku.
Creutzfeldt-Jakobova choroba
Priónová choroba, ktorá sa prejavuje demenciou, poruchami zraku a mozočku a poruchami pohybu s fatálnym koncom po 4-5 mesiacoch ochorenia pri klasickom variante Creutzfeldt-Jakobovej choroby a po (3-14 mesiacoch) pri novom variante Creutzfeldt-Jakobovej choroby. Inkubačná doba môže dosiahnuť 20 rokov. Možné sú rôzne cesty infekcie a príčiny ochorenia:
- pri konzumácii nedostatočne tepelne upravených živočíšnych produktov, ako je mäso a mozgy z kráv s bovinnou spongiformnou encefalopatiou;
- počas transplantácie tkaniva, ako je transplantácia rohovky, transfúzia krvi, použitie hormónov a iných biologicky aktívnych látok živočíšneho pôvodu, použitie katgutu, kontaminovaných alebo nedostatočne sterilizovaných chirurgických nástrojov, prosektúrne manipulácie;
- v prípade hyperprodukcie PrR a iných stavov, ktoré stimulujú proces premeny PrR' na PrR".
Ochorenie sa môže vyvinúť aj v dôsledku mutácie alebo inzercie v oblasti priónového génu. Familiárna povaha ochorenia je bežná kvôli genetickej predispozícii k Creutzfeldtovej-Jakobovej chorobe. Pri novom variante Creutzfeldtovej-Jakobovej choroby sa poruchy vyvíjajú v mladšom veku (priemerný vek 28 rokov), na rozdiel od klasického variantu (priemerný vek 65 rokov). Pri novom variante Creutzfeldtovej-Jakobovej choroby sa abnormálny priónový proteín hromadí nielen v centrálnom nervovom systéme, ale aj v lymforetikulárnych tkanivách vrátane mandlí.
Gerstmann-Sträussler-Scheinkerov syndróm
Dedičné priónové ochorenie sprevádzané demenciou, hypotóniou, poruchami prehĺtania (dysfágia), dyzartriou. Často má familiárny charakter. Inkubačná doba je od 5 do 30 rokov. Ochorenie sa vyskytuje vo veku 50 – 60 rokov, jeho trvanie sa pohybuje od 5 do 13 rokov.
Dedičná fatálna nespavosť
Autoimunitné ochorenie s progresívnou nespavosťou, sympatickou hyperreaktivitou (hypertenzia, hypertermia, hyperhidróza, tachykardia), tremorom, ataxiou, multiklonálnym syndrómom, halucináciami. Spánok je vážne narušený. S progresiou kardiovaskulárneho zlyhania nastáva smrť.
Škrabanie
Scrapie (z anglického scrape - škriabať) je priónové ochorenie oviec a kôz (svrab), ktoré sa prejavuje poškodením centrálneho nervového systému, progresívnymi poruchami pohybu, silným svrbením kože (svrab) a končí sa úhynom zvieraťa.
Bovinná spongiformná encefalopatia
Ochorenie hovädzieho dobytka charakterizované poškodením centrálneho nervového systému, zhoršenou koordináciou pohybov a nevyhnutnou smrťou zvieraťa. Epidémia choroby prvýkrát vypukla vo Veľkej Británii. Bola spojená s kŕmením zvierat mäsovou a kostnou múčkou obsahujúcou patologické prióny. Inkubačná doba sa pohybuje od 1,5 do 15 rokov. Najviac je infikovaný mozog, miecha a očné buľvy zvierat.
Laboratórna diagnostika priónových ochorení
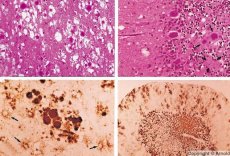
Počas diagnostiky sa zaznamenávajú spongiformné zmeny v mozgu, astrocytóza (glióza) a absencia zápalových infiltrátov. Mozog sa farbí na amyloid. V mozgovomiechovom moku sa detegujú proteínové markery priónových porúch mozgu (pomocou ELISA). Vykonáva sa genetická analýza priónového génu (PCR).
Prevencia priónových chorôb
Na dekontamináciu nástrojov a environmentálnych predmetov sa odporúča autoklávovanie (pri teplote 134 °C počas 18 minút; pri 121 °C počas 1 hodiny), spaľovanie, dodatočné ošetrenie bielidlom a roztokom NaCl s jednou kyselinou acetylsalicylovou počas 1 hodiny. Pre nešpecifickú profylaxiu boli zavedené obmedzenia týkajúce sa používania liekov živočíšneho pôvodu a zakázaná je produkcia hormónov hypofýzy živočíšneho pôvodu. Transplantácia tvrdej pleny mozgovej je obmedzená. Pri práci s tekutinami pacientov sa používajú gumené rukavice.