Lekársky expert článku

Nové publikácie
Neuroblastóm u detí: príčiny, diagnostika, liečba
Posledná kontrola: 04.07.2025

Všetok obsah iLive je lekársky kontrolovaný alebo kontrolovaný, aby sa zabezpečila čo najväčšia presnosť faktov.
Máme prísne smernice týkajúce sa získavania zdrojov a len odkaz na seriózne mediálne stránky, akademické výskumné inštitúcie a vždy, keď je to možné, na lekársky partnerské štúdie. Všimnite si, že čísla v zátvorkách ([1], [2] atď.) Sú odkazmi na kliknutia na tieto štúdie.
Ak máte pocit, že niektorý z našich obsahov je nepresný, neaktuálny alebo inak sporný, vyberte ho a stlačte kláves Ctrl + Enter.
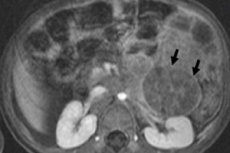
V detskej onkológii je jedným z najčastejších extrakraniálnych neoplaziem neuroblastóm u detí, čo je embryonálny malígny nádor neuroblastov neurálnej lišty, teda embryonálnych (nezrelých) nervových buniek sympatického nervového systému.
Epidemiológia
Podľa štatistík Medzinárodnej rizikovej skupiny pre neuroblastóm (INRG) predstavuje neuroblastóm približne 8 % všetkých onkologických ochorení u detí na celom svete a je tretím najčastejším ochorením po leukémii a mozgových nádoroch.
Podľa iných údajov predstavuje neuroblastóm približne 28 % všetkých druhov rakoviny u dojčiat. Viac ako tretina prípadov neuroblastómu je diagnostikovaná u detí mladších ako jeden rok; priemerný vek diagnózy je 19 – 22 mesiacov. Viac ako 90 % diagnostikovaných prípadov sa vyskytuje u detí vo veku od dvoch do piatich rokov (s prevahou chlapcov); vrchol incidencie sa pozoruje vo veku od dvoch do troch rokov a prípady u detí starších ako päť rokov tvoria menej ako 10 %.
Príčiny neuroblastómy
Pri štúdiu príčin neuroblastómu vedci dospeli k záveru, že tento nádor u detí vzniká v dôsledku sporadických genetických mutácií počas embryogenézy alebo skorého postnatálneho vývoja. Čo však spôsobuje tieto génové zmeny, nie je známe, pretože nebol identifikovaný žiadny vplyv teratogénnych faktorov prostredia.
Tieto nádory sa môžu vyskytnúť kdekoľvek, vrátane mediastina, krku, brucha, nadobličiek, obličiek, chrbtice a panvy.
V zriedkavých prípadoch môže byť neuroblastóm u dojčiat spojený s dedičnou mutáciou. Konkrétne ide o mutáciu v géne membránového proteínu CD246 na chromozóme 2 - enzýmu tyrozínkinázy ALK, ktorý zabezpečuje medzibunkovú komunikáciu a hrá dôležitú úlohu vo fungovaní nervového systému; v géne proteínu PHOX2B (na chromozóme 4), ktorý sa podieľa na dozrievaní nervových buniek.
Neuroblastóm môže byť tiež spojený s detskou neurofibromatózou typu 1,Beckwithovým-Wiedemannovým syndrómom a hyperinzulinemickou hypoglykémiou (nezidioblastózna pankreatitída).
Rizikové faktory
Dnes sa dedičnosť uznáva ako rizikový faktor pre rozvoj neuroblastómu u detí - prítomnosť tohto nádoru v rodinnej anamnéze, ako aj vrodené anomálie spojené s génovými mutáciami počas vnútromaternicového vývoja. Platí to najmä pre prípady vzniku viacerých novotvarov v rôznych orgánoch.
Výskumníci neidentifikovali žiadny z exogénnych faktorov, ktoré zvyšujú riziko tohto nádoru.
Patogenézy
Mechanizmus vývoja neuroblastómov je spôsobený poruchami diferenciácie a dozrievania buniek neurálnej lišty – bilaterálnych bunkových línií, ktoré sa tvoria na okrajoch neurálnej trubice z ektodermálnej zárodočnej vrstvy ľudského embrya. Tieto bunky migrujú (presúvajú sa) a diferencujú sa na mnoho typov buniek: senzorické a autonómne neuróny, neuroendokrinné bunky a bunky drene nadobličiek, bunky kraniofaciálnej chrupavky a kostí, ako aj pigmentové bunky.
Pri neuroblastóme migrované neuroblasty nedozrievajú, ale pokračujú v raste a delení, čím vytvárajú nádor. A patogenéza jeho vzniku je spojená s nasledujúcimi génovými mutáciami:
- s duplikáciou časti chromozómovej sekvencie alebo duplikáciou segmentov génu LMO1 na chromozóme 11, kódujúceho proteín RBTN1 v bunkách nervového hrebeňa embrya;
- so zmenou počtu kópií génu NBPF10 na chromozóme 1q21.1, kódujúcom proteín DUF1220, ktorý riadi proliferáciu ľudských nervových kmeňových buniek. Tieto poruchy vedú buď k duplikácii tohto chromozómu, alebo k jeho delécii – absencii časti DNA;
- so zmenami v géne tumor supresora ATRX (na chromozóme Xq21.1);
- s prítomnosťou ďalších kópií (amplifikácia) génu transkripčného faktora N-Myc na chromozóme 2, ktorý kóduje jeden z transkripčných faktorov (proteín viažuci DNA), ktorý reguluje aktivitu iných génov a riadi proliferáciu prekurzorových buniek počas tvorby proteínov pre tvorbu tkanív a orgánov plodu. Amplifikácia tohto génu ho mení na onkogén, čo vyvoláva narušenie bunkového cyklu, zvýšenú proliferáciu buniek a tvorbu nádorov.
Príznaky neuroblastómy
Prvé príznaky neuroblastómu sú nešpecifické a môžu zahŕňať stratu chuti do jedla (a úbytok hmotnosti), únavu pri kŕmení, horúčku a bolesť kĺbov.
Klinické príznaky závisia od umiestnenia primárneho nádoru a prítomnosti metastáz (ktoré sa vyskytujú v 60 – 73 % prípadov).
Primárny neuroblastóm je veľmi často lokalizovaný v dreni nadobličiek, ktorá má podobný pôvod ako nervové bunky. U detí mladších ako jeden rok je neuroblastóm nadobličiek diagnostikovaný v 35 – 40 % prípadov. Medzi jeho príznaky patrí bolesť brucha, horúčka, úbytok hmotnosti, bolesť kostí, anémia alebo sprievodný Pepperov syndróm: difúzne poškodenie pečene s ťažkou hepatomegáliou a syndrómom respiračnej tiesne.
Retroperitoneálny neuroblastóm alebo retroperitoneálny neuroblastóm u detí, ako rastie, začína tlačiť na močový mechúr alebo črevá, čo môže spôsobiť problémy s močením alebo defekáciou, opuch nôh (u chlapcov opuchuje miešok).
Neuroblastóm mediastina u detí (mediastinálny neuroblastóm) často tlačí na hornú dutú žilu, čo môže spôsobiť opuch tváre, krku, rúk a hornej časti hrudníka (s modročervenou pokožkou a podkožnými uzlíkmi). Objavuje sa kašeľ a sipot, problémy s dýchaním (dýchavičnosť) alebo problémy s prehĺtaním (dysfágia); zväčšené lymfatické uzliny sa pozorujú na krku, nad kľúčnou kosťou a v podpazuší.
Šírenie nádorových buniek do kostnej drene vedie k anémii, trombocytopénii a leukopénii so sklonom ku krvácaniu.
A s metastázami v periorbitálnej oblasti sa okolo očí objavujú tmavé kruhy alebo modriny. Takýto nádor môže tiež spôsobiť bolesti hlavy a závraty, exoftalmiu (vydutie očných buliev) a v dôsledku kompresie nervových zakončení - poklesnutie očných viečok (ptóza) a zmenšenie veľkosti zreníc (mióza).
Abdominálny neuroblastóm alebo neuroblastóm brušnej dutiny u detí vedie k tvorbe hmatateľných tesnení v brušnej dutine, jej nadúvaniu, strate chuti do jedla, zápche a zvýšenému krvnému tlaku. Nádor tlačiaci na miechu alebo nervový koreň môže viesť k znecitliveniu a slabosti končatín, neschopnosti stáť, plaziť sa alebo chodiť. Ak sú postihnuté kosti, môže sa vyskytnúť bolesť kostí.
V prípade nádoru 3. – 4. štádia v brušnej dutine s poškodením lymfatických uzlín môžu nádorové bunky preniknúť do renálneho parenchýmu a potom sa u detí vyvinie rozsiahly neuroblastóm obličiek, čo vedie k narušeniu ich funkcií.
Etapy
- Neuroblastóm 1. štádia je primárny nádor, ktorý je lokalizovaný a izolovaný v jednej oblasti tela; lymfatické uzliny na oboch stranách nie sú postihnuté.
- Neuroblastóm v štádiu 2. V štádiu 2A je primárny nádor obmedzený na jednu oblasť, ale je veľký; nie sú postihnuté bilaterálne lymfatické uzliny. V štádiu 2B sú lymfatické uzliny na strane tela, kde sa nádor nachádza, pozitívne na metastázy.
- Neuroblastóm v 3. štádiu: primárny nádor prechádza cez miechu alebo stredovú čiaru tela, v lymfatických uzlinách sa nachádzajú jednostranné alebo obojstranné metastázy.
- Neuroblastóm štádium 4: nádor sa rozšíril do vzdialených lymfatických uzlín, kostnej drene, kostí, pečene alebo iných orgánov. Štádium 4S sa zisťuje u detí mladších ako jeden rok s lokalizovaným primárnym nádorom s disemináciou do kože, pečene alebo kostnej drene.
Medzinárodný systém klasifikácie rizika neuroblastómu (INRGSS)
INRGSS používa rizikové faktory definované zobrazovaním (IDRF), čo sú faktory pozorované pri zobrazovacích vyšetreniach, ktoré môžu znamenať, že odstránenie nádoru bude ťažšie.
INRGSS rozdeľuje neuroblastómy do 4 štádií:
- L1: Nádor sa nerozšíril z miesta, kde vznikol, a neprerástol do životne dôležitých štruktúr. Je obmedzený na jednu časť tela, ako je krk, hrudník alebo brucho.
- L2: Nádor sa nerozšíril (nemetastázoval) ďaleko od miesta, kde začal (napríklad mohol rásť z ľavej strany brucha na ľavú stranu hrudníka), ale má aspoň jeden IDRF.
- M: Nádor metastázoval do vzdialenej časti tela (s výnimkou nádorov v štádiu sklerózy multiplex).
- MS: Metastatické ochorenie u detí mladších ako 18 mesiacov, pri ktorom sa rakovina rozšírila iba do kože, pečene a/alebo kostnej drene.
Komplikácie a následky
Neuroblastóm sa vyznačuje komplikáciami a následkami, ako sú:
- šírenie (metastázy) do lymfatických uzlín, kostnej drene, pečene, kože a kostí;
- kompresia miechy (ktorá môže spôsobiť bolesť a viesť k paralýze);
- rozvoj paraneoplastického syndrómu (v dôsledku pôsobenia určitých chemických látok vylučovaných nádorom, ako aj antigénu disialogangliozidu GD2 exprimovaného jeho bunkami), ktorý sa prejavuje rýchlymi mimovoľnými pohybmi očí, zhoršenou koordináciou, svalovými kŕčmi a hnačkou;
- relapsy po ukončení primárnej terapie (ako ukazuje klinická prax, vysokorizikové neuroblastómy majú relaps v 50 % prípadov).
Diagnostika neuroblastómy
Diagnóza podozrenia na neuroblastóm u dieťaťa vyžaduje vyšetrenie, laboratórne testy a zobrazovacie metódy.
Vykonávajú sa krvné a močové testy na katecholamíny (norepinefrín a dopamín) a kyseliny homovanilovej alebo vanilylmandľovej (vznikajúce počas metabolizmu týchto hormónov); krvný test na neurospecifickú enolázu, enzýmovo-imunoanalýza (ELISA) krvného séra a analýza kostnej drene (jej vzorka sa odoberá aspiračnou punkciou). Vykoná sa test DNA na stanovenie mutácií a biopsia na cytomorfologické vyšetrenie nádorového tkaniva.
Po odobratí vzoriek biopsie sa tieto odošlú do laboratória, kde ich pod mikroskopom vyšetrí patológ (lekár so špeciálnym vzdelaním v oblasti identifikácie rakovinových buniek). Na vzorkách sa často vykonávajú aj špeciálne laboratórne testy, aby sa zistilo, či ide o neuroblastóm.
Ak ide o neuroblastóm, laboratórne testy môžu tiež pomôcť určiť, ako rýchlo môže nádor rásť alebo sa šíriť, ako aj aké liečebné postupy by mohli fungovať najlepšie.
Inštrumentálna diagnostika vizualizuje novotvar pomocou ultrazvuku, röntgenu, MRI alebo CT, PET so zavedením 18F-fluórdeoxyglukózy alebo MIBG skenovaním - scintigrafiou s metajódbenzylguanidínom. [ 1 ]
Odlišná diagnóza
Diferenciálna diagnostika zahŕňa benígny ganglioneuróm, ganglioneuroblastóm, rabdomyosarkóm a nefroblastóm.
Komu sa chcete obrátiť?
Liečba neuroblastómy
Pri neuroblastóme závisí liečba od rizikovej skupiny pacienta (štádium nádorového procesu), lokalizácie nádoru, genomických znakov nádorových buniek a veku dieťaťa. A môže zahŕňať monitorovanie, chirurgický zákrok, chemoterapiu, rádioterapiu, imunoterapiu a transplantáciu hematopoetických kmeňových buniek.
Neoadjuvantná alebo adjuvantná (predoperačná alebo pooperačná) chemoterapia neuroblastómu u detí, rovnako ako akákoľvek chemoterapia rakoviny, sa podáva v cykloch: liek sa podáva niekoľko dní po sebe, po ktorých nasleduje prestávka na zotavenie tela. Cykly sa zvyčajne opakujú každé tri až štyri týždne.
Používajú sa nasledujúce lieky (a ich kombinácie): cyklofosfamid, cisplatina alebo karboplatina, doxorubicín (adriamycín), vinkristín, etopozid.
Medzi bežné vedľajšie účinky chemoterapie patrí vypadávanie vlasov, strata chuti do jedla, únava, nevoľnosť a vracanie, afty v ústach, hnačka alebo zápcha. Chemoterapia môže poškodiť kostnú dreň a spôsobiť zníženie počtu krviniek.
Cielená imunoterapia (zameraná na nádorový antigén GD2) využíva lieky zo skupiny monoklonálnych protilátok (anti-GD2 MAb) Dinutuximab (Unituxin) a Naxitamab. Podávajú sa intravenózne dlhodobou infúziou v kombinácii s faktorom stimulujúcim kolónie granulocytov a makrofágov (cytokín GM-CSF) a interleukínom-2.
Medzi vedľajšie účinky týchto liekov patrí bolesť (často veľmi silná), znížený krvný tlak, zvýšená srdcová frekvencia, dýchavičnosť (s možným opuchom dýchacích ciest), zvýšená teplota, nevoľnosť, vracanie a hnačka, zmeny v bunkovom a minerálnom zložení krvi.
Aby sa znížilo riziko recidívy rakoviny po vysokodávkovej chemoterapii a transplantácii kmeňových buniek, deti s vysokorizikovým neuroblastómom sa liečia systémovými retinoidmi, kyselinou 13-cis-retínovou (izotretinoínom). [ 2 ]
Chirurgická liečba neuroblastómu – odstránenie nádoru, napríklad otvorená adrenalektómia alebo laparoskopická resekcia neuroblastómu nadobličiek; lymfektómia (odstránenie postihnutých lymfatických uzlín) atď. [ 3 ]
Pri vysokorizikovom neuroblastóme sa môže použiť rádioterapia.[ 4 ]
Prevencia
Vzhľadom na príčiny neuroblastómu u detí môže byť jediným preventívnym opatrením genetické poradenstvo pri plánovaní tehotenstva. Treba však mať na pamäti, že tento nádor je spojený s dedičnými mutáciami iba v 1-2% prípadov.
Predpoveď
Infantilný neuroblastóm má schopnosť spontánnej regresie.
Prognostické markery
- Vysokorizikové nádory, ako aj neuroblastóm u detí všetkých vekových skupín a všetkých štádií (okrem štádia 4S) – so zvýšenou expresiou génu N-MYC a amplifikáciou onkogénu N-Myc – majú nepriaznivú prognózu, ktorá ovplyvňuje dĺžku života.
- Nádorové bunky, ktorým chýbajú určité časti chromozómov 1 alebo 11 (známe ako delécie 1p alebo 11q), majú horšiu prognózu. S horšou prognózou je spojená aj prítomnosť ďalšej časti chromozómu 17 (zvýšenie 17q).
- Neuroblastómové bunky s veľkým množstvom DNA majú lepšiu prognózu, najmä u detí mladších ako 2 roky.
- Neuroblastómy, ktoré majú viac neurotrofínových receptorov, najmä receptor nervového rastového faktora TrkA, majú lepšiu prognózu.
Prežitie podľa rizikovej skupiny detskej onkológie (COG)
- Skupina s nízkym rizikom: Deti v skupine s nízkym rizikom majú 5-ročnú mieru prežitia vyššiu ako 95 %.
- Stredne riziková skupina: Deti v stredne rizikovej skupine majú 5-ročnú mieru prežitia 90 % až 95 %.
- Vysoko riziková skupina: Deti vo vysoko rizikovej skupine majú 5-ročnú mieru prežitia približne 50 %.
Približne 15 % úmrtí na rakovinu v detstve je spôsobených neuroblastómom. Šanca na dlhodobé prežitie pri tomto vysoko rizikovom zhubnom nádore nepresahuje 40 %. Celková päťročná miera prežitia je 67 – 74 %, 43 % vo vekovej skupine od jedného do štyroch rokov a viac ako 80 % pri neuroblastóme diagnostikovanom v prvom roku života.
Использованная литература